Galactosylceramidase
IntroductionGalactosylceramidase (or galactocerebrosidase) is a hydrolase [1] that removes galactose from galactosylceramide and other sphingolipids[2]. Galactosylceramidase in humans is encoded by the gene GALC, and mutations in this gene are associated with Krabbe disease, or globoid cell leukodystrophy[3].
See also Lipid signaling. StructureX-ray diffraction data () from mouse models indicates that GALC is an estimated 77 kDa monomer consisting of a 656 residues, which form a containing 12 α-helices and 41 β-strands[1]. Each β-strand contains three to eleven residues. The enzyme contains three major domains: a central triosephosphate isomerase (TIM) barrel, a β-sandwich domain, and a lectin domain[3]. The TIM barrel is composed of eight parallel β-strands surrounded by α-helices and a single disulfide bridge between residues Cys 271 and Cys 378, while the β-sandwich is composed of two twisted β-sheets. The lectin domain is characterized by a calcium ion that is bound in pentagonal bipyramidal configuration[3]. The substrate-binding site of GALC is mostly comprised of the central TIM barrel, however loops from the other two domains also appear to contribute to the binding pocket. Residues that appear to interact with the substrate by hydrogen bonding include Gly48, Thr93, Trp135, Asn181, Glu182, Glu258, Ser261, and Arg380. Of particular interest are the residues Glu258 and Glu182, which have been proposed as the active site nucleophile and proton donor respectively, due to the average distance between the carboxyl oxygens being consistent with the retaining mechanism of glycosidic bond hydrolysis.[3] FunctionThe molecular function of galactosylceramidase is hydrolysis of a O-glycosyl bond to remove galactose from Ceramide and other sphingolipids. The cellular function is the maintenance of a functional hematopoietic stem/progenitor cell niche by contributing to the control of the intracellular content of key sphingolipids[4].  DiseaseDefects in this enzyme cause a lysosomal storage disorder known in humans as Krabbe disease (or globoid cell leukodystrophy). Krabbe disease is a neurodegenerative disorder characterized by widespread demyelination caused by reduced or mutated function of GALC[3]. The deficiency of GALC leads to the accumulation of the neurotoxic metabolite 1-β-d-galactosylsphingosine (psychosine) in the central nervous system. Psychosine causes the destruction of epithelial actin structures and is toxic to oligodendrocytes[5][6], by causing lipid raft clustering which leads to defective signal transduction[7]. Besides the defective recruitment of signal molecules to lipid rafts, there is also impairment in the early steps of endocytosis and vesicle transport[7]. Defects in the retrograde axonal transport is correlated with decreased amounts of dynein, unusual levels of post-translational tubulin modifications, and microtubule instability[7]. GALC deficiency also causes the accumulation of lipids in "globoid" macrophages, where the medical name for the disease originated[6]. A common authentic model for this disease is the twitcher mouse model[5]. The only treatment currently available is an experiemental hematopoietic stem cell transplant, and gene therapies and enzyme replacements are still being researched[6].
|
| ||||||||||||||||||||||||
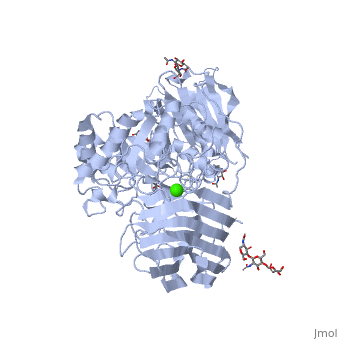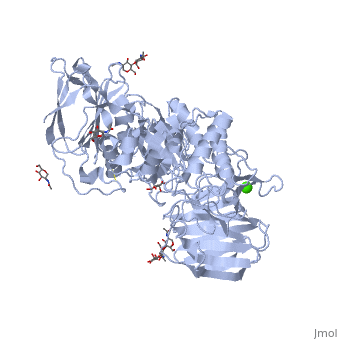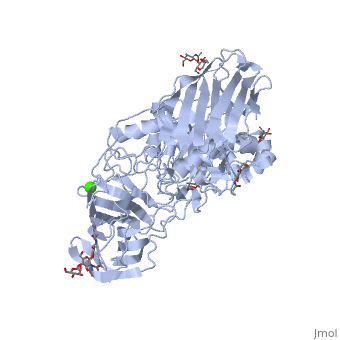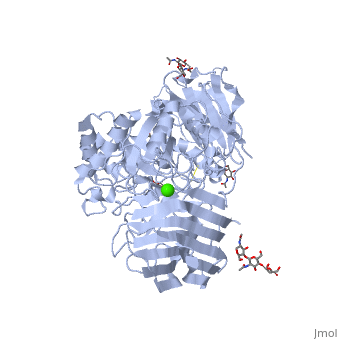
Krabbe disease is a devastating neurodegenerative disease characterized by widespread demyelination that is caused by defects in the enzyme galactocerebrosidase (GALC). Disease-causing mutations have been identified throughout the GALC gene. However, a molecular understanding of the effect of these mutations has been hampered by the lack of structural data for this enzyme. Here we present the crystal structures of GALC and the GALC-product complex, revealing a novel domain architecture with a previously uncharacterized lectin domain not observed in other hydrolases. All three domains of GALC contribute residues to the substrate-binding pocket, and disease-causing mutations are widely distributed throughout the protein. Our structures provide an essential insight into the diverse effects of pathogenic mutations on GALC function in human Krabbe variants and a compelling explanation for the severity of many mutations associated with fatal infantile disease. The localization of disease-associated mutations in the structure of GALC will facilitate identification of those patients that would be responsive to pharmacological chaperone therapies. Furthermore, our structure provides the atomic framework for the design of such drugs.
Insights into Krabbe disease from structures of galactocerebrosidase., Deane JE, Graham SC, Kim NN, Stein PE, McNair R, Cachon-Gonzalez MB, Cox TM, Read RJ, Proc Natl Acad Sci U S A. 2011 Sep 13;108(37):15169-73. Epub 2011 Aug 29. PMID:21876145
From MEDLINE®/PubMed®, a database of the U.S. National Library of Medicine.
LigandsLigands
- calcium ion
- N-acetyl-D-glucosamine

InhibitorsInhibitors
Because Krabbe disease is caused by a decrease in galactosylceramidase activity, there are no drug-protein interactions to explore. However some natural inhibitory molecules in humans include:
- 6-hexadecanoylamino-4-methylbelliferyl-beta-D-galactopyranoside, competitive inhibition
- D-galactose
- galactonyl hydrazide
- lactose
- N-(6-aminohexyl)-D-galactoside
- taurocholate (at high concentrations above 0.3% w/v)[8]
QuizQuiz
3D Structures of galactosylceramidase3D Structures of galactosylceramidase
Updated on 15-January-2023
3zr5 – mGC – mouse
3zr6, 4cce – mGC + galactose
4ccc – mGC + nitrophenyl galactopyranose
4ccd – mGC + galactal
6y6s, 6y6t – mGC + galacto-noeurostegine
4ufh, 4ufi, 4ufj, 4ufl, 4ufk, 4ufm – mGC + azasugar inhibitor
5nxb – mGC + saposin A
ReferencesReferences
- ↑ 1.0 1.1 RCSB Protein Data Bank - RCSB PDB - 3ZR5 Structure Summary. (n.d.). RCSB Protein Data Bank - RCSB PDB - 3ZR5 Structure Summary. Retrieved June 3, 2014, from www.rcsb.org DOI:10.2210/pdb3zr5/pdb
- ↑ Zizioli D, Guarienti M, Tobia C, Gariano G, Borsani G, Bresciani R, Ronca R, Giacopuzzi E, Preti A, Gaudenzi G, Belleri M, Di Salle E, Fabrias G, Casas J, Ribatti D, Monti E, Presta M. Molecular cloning and knockdown of galactocerebrosidase in zebrafish: new insights into the pathogenesis of Krabbe's disease. Biochim Biophys Acta. 2014 Apr;1842(4):665-75. doi: 10.1016/j.bbadis.2014.01.008., Epub 2014 Jan 24. PMID:24463171 doi:http://dx.doi.org/10.1016/j.bbadis.2014.01.008
- ↑ 3.0 3.1 3.2 3.3 3.4 Deane JE, Graham SC, Kim NN, Stein PE, McNair R, Cachon-Gonzalez MB, Cox TM, Read RJ. Insights into Krabbe disease from structures of galactocerebrosidase. Proc Natl Acad Sci U S A. 2011 Sep 13;108(37):15169-73. Epub 2011 Aug 29. PMID:21876145 doi:10.1073/pnas.1105639108
- ↑ Visigalli I, Ungari S, Martino S, Park H, Cesani M, Gentner B, Sergi Sergi L, Orlacchio A, Naldini L, Biffi A. The galactocerebrosidase enzyme contributes to the maintenance of a functional hematopoietic stem cell niche. Blood. 2010 Sep 16;116(11):1857-66. doi: 10.1182/blood-2009-12-256461. Epub 2010 , May 28. PMID:20511539 doi:http://dx.doi.org/10.1182/blood-2009-12-256461
- ↑ 5.0 5.1 Belleri M, Ronca R, Coltrini D, Nico B, Ribatti D, Poliani PL, Giacomini A, Alessi P, Marchesini S, Santos MB, Bongarzone ER, Presta M. Inhibition of angiogenesis by beta-galactosylceramidase deficiency in globoid cell leukodystrophy. Brain. 2013 Sep;136(Pt 9):2859-75. doi: 10.1093/brain/awt215. PMID:23983033 doi:http://dx.doi.org/10.1093/brain/awt215
- ↑ 6.0 6.1 6.2 Kohlschutter A. Lysosomal leukodystrophies: Krabbe disease and metachromatic leukodystrophy. Handb Clin Neurol. 2013;113:1611-8. doi: 10.1016/B978-0-444-59565-2.00029-0. PMID:23622382 doi:http://dx.doi.org/10.1016/B978-0-444-59565-2.00029-0
- ↑ 7.0 7.1 7.2 Teixeira CA, Miranda CO, Sousa VF, Santos TE, Malheiro AR, Solomon M, Maegawa GH, Brites P, Sousa MM. Early axonal loss accompanied by impaired endocytosis, abnormal axonal transport, and decreased microtubule stability occur in the model of Krabbe's disease. Neurobiol Dis. 2014 Jun;66:92-103. doi: 10.1016/j.nbd.2014.02.012. Epub 2014 Mar , 6. PMID:24607884 doi:http://dx.doi.org/10.1016/j.nbd.2014.02.012
- ↑ EC 3.2.1.46 - galactosylceramidase. (n.d.). Information on. Retrieved June 3, 2014, from www.brenda-enzymes.org