Fatty acid amide hydrolase
This page, as it appeared on May 3, 2014, was featured in this article in the journal Biochemistry and Molecular Biology Education.
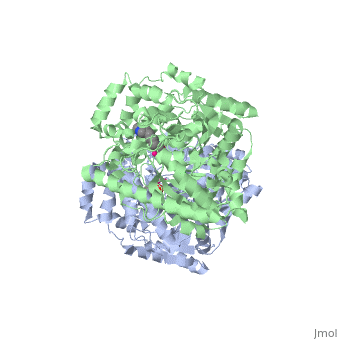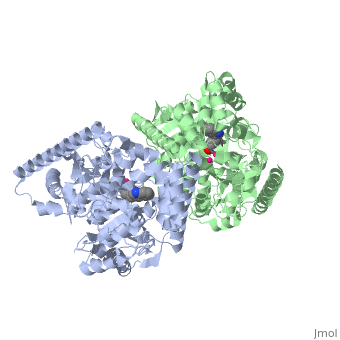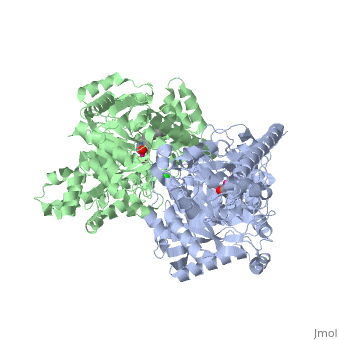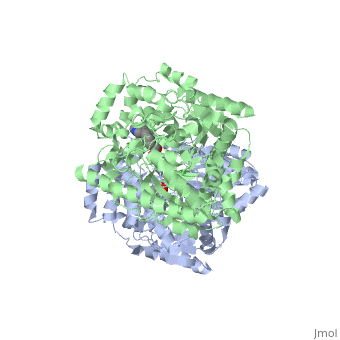
IntroductionFatty acid amide hydrolase (FAAH) degrades fatty acid amides (FAAs) to terminate their signaling activity [1]. A serine hydrolase from the Amidase signature superfamily of enzymes (other amidases), FAAH degrades endocannabinoid signaling lipids, molecules associated with pain relief [2]. Because endocannabinoids are lipid molecules, they cannot be compartmentalized in vesicles (the degradation method for other neurotransmitters) and must instead be degraded in the bilayer of the cell membrane. FAAH is an integral membrane protein that degrades FAAs as they enter the membrane bilayer, allowing the cell to terminate the activity of signaling molecules that cannot be contained within a vesicle for degredation [1]. Current FAAH research aims to find inhibitors for the enzyme, which would prolong the pain alleviation provided by endocannabinoid molecules [2].  General Structure InformationCrystal structures of FAAH show that the enzyme is a (monomers in different colors) in solution, with each subunit having a mass of 63 kD [1]. The protein's of 11 strands (blue) is surrounded by 24 (green). The enzyme is embedded in the cell membrane to catch the lipid signaling molecules that can diffuse through membranes; the hydrolase has a membrane binding cap, a consisting of alpha helices 18 and 19. These helices present hydrophobic amino acid residues that help FAAH interact with the hydrophobic region of the lipid bilayer [1]. Specifically, (black) interact with the hydrophobic region of the membrane to anchor the enzyme in the lipid bilayer. The FAAH structure shows an entry channel leading from the lipid bilayer to the enzyme's active site, providing a path for endocannabinoids to enter the hydrolase. This is amphipathic to accommodate the entire lipid signaling molecule. Hydrophobic amino acid residues (green) interact with the lipid signaling molecules' nonpolar tails, while charged R486 and D403 residues (black) in the entry channel accommodate the polar head groups. In addition, FAAH possesses an exit channel leading from the active site to the cell's cytoplasm, allowing the release of polar compounds released from lipid cleavage and the entry of water molecules necessary for the FAAH mechanism to proceed [1]. Different inhibitors have been designed to learn more about species selectivity [3] and binding flexibility [4] in FAAH. For example, the (red) is the inhibitor used on a humanized rat FAAH protein. Although PF-750 showed preference for human FAAH, rat FAAH is easier to express; this demonstrates species selectivity of inhibitors [3]. Membrane Access ChannelThe of FAAH reveals two equivalent openings (, ) directly accessible by the inner layer of the lipid bilayer.[1] These (MAC) are made up of three flaps and two intrusions which collectively form the entry way for the aliphatic binding of the amide lipid substrate. Flaps 1 and 2 envelope the middle and backside of the anandamide mimic, and are locked together by a between Arg486 and Asp403. Flap 2 contains a very that partially covers the opening with Phe432. This binding cap clings to the cell's hydrophobic inner membrane and uses a multitude of to lure out partitioned anandamide by its narrow partial negative charge. The catalytic site is defined by the catalytic triad: the contributes the nucleophilic S241, loop 3 contributes the neighboring upon forming the very top of the membrane access channel, and a fourth loop contributes the deprotonating . The membrane access channel leads to the active site, which is flanked by both the and the acyl chain binding pocket. The cytosolic port is a lengthy, flexible loop that leads directly into the cytoplasm, allowing the deacylated amine to enter the cell, while the acyl chain binding pocket is a space formed by several of the loops that accommodates the substrates polar head.  Catalytic TriadMutagenesis and inhibitor studies have shown that FAAH has a , consisting of Ser241, Ser217, and Lys142 [1]. Ser-Ser-Lys catalytic triads are not often seen in hydrolases, making FAAH an enzyme of interest for additional research to better determine how proteins with this catalytic triad function. Ser241 acts as the catalytic nucleophile for the cleavage of amide bonds (Figure 2) [1]. Inhibitors are able to inactivate the catalytic triad by providing a substrate containing a leaving group, such as aniline, that is a more favorable leaving group than the Ser241 hydroxyl group. With the serine bound to the carbonyl carbon, FAAH is no longer able to accommodate any more substrates [3]. FAAH also requires two water molecules in its active site to properly position and cleave amide bonds. One water molecule (W1) deacylates the substrate, and the other (W2) helps coordinate W1 through the catalytic K142 (Figure 4) [5] .  Relationship to other proteinsThe hydrolytic water molecules important to FAAH's function suggest an evolutionary relationship of this hydrolase to other enzymes. The structures of other serine hydrolases also display a catalytic water molecule in their active sites. Because hydrolases that are non-homologous to FAAH also require a water molecule to cleave bonds, a functional convergance has inferred between amidase signature enzymes (such as FAAH) and other classes of serine proteases [5].  This evidence of convergent evolution between FAAH and other amidase signature enzymes supports the Bürgi-Dunitz theory. The Bürgi-Dunitz theory proposes that nucleophiles tend to follow a specific trajectory when attacking a carbonyl, resulting in many enzyme mechanisms having the same angle between an incoming nucleophile and the carbonyl it attacks (FAAH bound to inhibitor). Water molecules in the active sites of enzymes are specifically positioned to force the nucleophile to approach at the exact "Bürgi-Dunitz angle" of 107°. The determination that FAAH also has water molecules in its active site, helping the nucleophile to attack the amide carbonyl at a specific angle, adds additional support to the Bürgi-Dunitz theory [5]. ApplicationsThe human nervous system has several types of chemical messengers, including amino acids, lipids, peptide hormones, and monoamines [1]. FAAH primarily degrades anandamide (AEA), a naturally-occurring signaling lipid that functions in the brain. AEA brings pain relief to the body. Inhibiting FAAH would likely sustain AEA signaling, leading to prolonged pain relief and decreased inflammation [2].  FAAH plays a role in endocannabinoid signaling that has intriguing potential as a drug target. This signaling system consists of endocannabinoid ligands (such as AEA), two G protein-coupled receptors (CB1 and CB2), and the enzymes that synthesize and degrade (such as FAAH) the signaling lipids. Previous research has explored the potential of regulating endocannabinoid signaling through the CB1 and CB2 receptors. However, molecules found to activate these receptors (such as tetrahydrocannabinol (THC), the main psychoactive ingredient of marijuana), while providing the intended pain relief, also produce many undesirable side effects, such as decreased cognition and motor control. On the other hand, research involving FAAH inhibitors has shown that blocking this part of the pathway reduces pain without the unwanted side effects seen through CB1/CB2 activation. Thus, exploring the possibility of using FAAH inhibition to decrease pain relief with minimal side effects could lead to new pain treatment solutions. For example, the PF-3845 inhibitorof FAAH is extremely selective for the enzyme and raises AEA levels for a prolonged duration of time. Therefore, this inhibitor could be explored as a possible pharmaceutical product to target FAAH when extended pain relief is desired [2]. 3D Structures of fatty acid amide hydrolaseUpdated on 04-May-2025 3qj8 – rFAAH - rat 1mt5, 3qj9, 3qkv, 4hbp – rFAAH + inhibitor
|
| ||||||||||
ReferencesReferences
- ↑ 1.0 1.1 1.2 1.3 1.4 1.5 1.6 1.7 1.8 Bracey MH, Hanson MA, Masuda KR, Stevens RC, Cravatt BF. Structural adaptations in a membrane enzyme that terminates endocannabinoid signaling. Science. 2002 Nov 29;298(5599):1793-6. PMID:12459591 doi:10.1126/science.1076535
- ↑ 2.0 2.1 2.2 2.3 Ahn K, Johnson DS, Mileni M, Beidler D, Long JZ, McKinney MK, Weerapana E, Sadagopan N, Liimatta M, Smith SE, Lazerwith S, Stiff C, Kamtekar S, Bhattacharya K, Zhang Y, Swaney S, Van Becelaere K, Stevens RC, Cravatt BF. Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem Biol. 2009 Apr 24;16(4):411-20. PMID:19389627 doi:10.1016/j.chembiol.2009.02.013
- ↑ 3.0 3.1 3.2 3.3 Mileni M, Johnson DS, Wang Z, Everdeen DS, Liimatta M, Pabst B, Bhattacharya K, Nugent RA, Kamtekar S, Cravatt BF, Ahn K, Stevens RC. Structure-guided inhibitor design for human FAAH by interspecies active site conversion. Proc Natl Acad Sci U S A. 2008 Sep 2;105(35):12820-4. Epub 2008 Aug 27. PMID:18753625
- ↑ Mileni M, Garfunkle J, DeMartino JK, Cravatt BF, Boger DL, Stevens RC. Binding and inactivation mechanism of a humanized fatty acid amide hydrolase by alpha-ketoheterocycle inhibitors revealed from cocrystal structures. J Am Chem Soc. 2009 Aug 5;131(30):10497-506. PMID:19722626 doi:10.1021/ja902694n
- ↑ 5.0 5.1 5.2 Mileni M, Kamtekar S, Wood DC, Benson TE, Cravatt BF, Stevens RC. Crystal structure of fatty acid amide hydrolase bound to the carbamate inhibitor URB597: discovery of a deacylating water molecule and insight into enzyme inactivation. J Mol Biol. 2010 Jul 23;400(4):743-54. Epub 2010 May 21. PMID:20493882 doi:10.1016/j.jmb.2010.05.034
Similar Proteopedia PagesSimilar Proteopedia Pages
Student ContributorsStudent Contributors
- Rachel Erkilla
- Melissa Jones
- Daniel Lange
- Carter Sharp