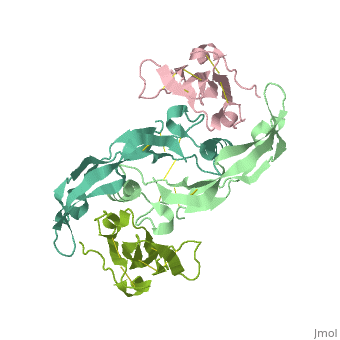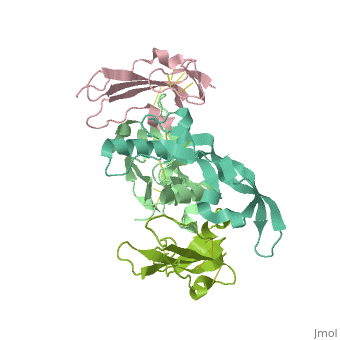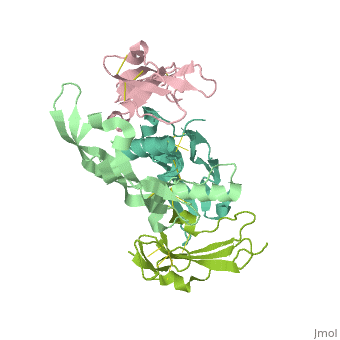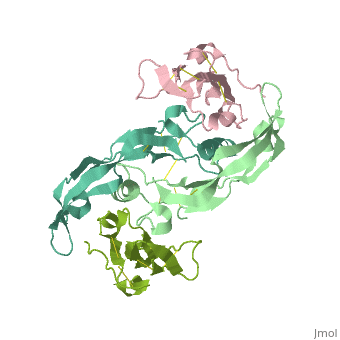3evs
Crystal structure of the GDF-5:BMP receptor IB complex.Crystal structure of the GDF-5:BMP receptor IB complex.
Structural highlights
DiseaseGDF5_HUMAN Defects in GDF5 are the cause of acromesomelic chondrodysplasia Grebe type (AMDG) [MIM:200700. Acromesomelic chondrodysplasias are rare hereditary skeletal disorders characterized by short stature, very short limbs, and hand/foot malformations. The severity of limb abnormalities increases from proximal to distal with profoundly affected hands and feet showing brachydactyly and/or rudimentary fingers (knob-like fingers). AMDG is an autosomal recessive form characterized by normal axial skeletons and missing or fused skeletal elements within the hands and feet.[1] Defects in GDF5 are the cause of acromesomelic chondrodysplasia Hunter-Thompson type (AMDH) [MIM:201250. AMDH is an autosomal recessive form of dwarfism. Patients have limb abnormalities, with the middle and distal segments being most affected and the lower limbs more affected than the upper. AMDH is characterized by normal axial skeletons and missing or fused skeletal elements within the hands and feet. Defects in GDF5 are the cause of brachydactyly type C (BDC) [MIM:113100. BDC is an autosomal dominant disorder characterized by an abnormal shortness of the fingers and toes. Note=Some BDC patients with GDF5 mutations also manifest clinical features of ASPED angel-shaped phalango-epiphyseal dysplasia (ASPED), an autosomal dominant skeletal abnormality characterized by a typical angel-shaped phalanx, brachydactyly, specific radiological findings, abnormal dentition, hip dysplasia, and delayed bone age. This suggests that BDC and ASPED are part of the same clinical spectrum (PubMed:22828468).[2] [3] Defects in GDF5 are the cause of Du Pan syndrome (DPS) [MIM:228900; also known as fibular hypoplasia and complex brachydactyly. Du Pan syndrome is a rare autosomal recessive condition characterized by absence of the fibulae and severe acromesomelic limb shortening with small, non-functional toes. Although milder, the phenotype resembles the autosomal recessive Hunter-Thompson and Grebe types of acromesomelic chondrodysplasia.[4] [5] [6] Defects in GDF5 are a cause of symphalangism proximal syndrome (SYM1) [MIM:185800. SYM1 is characterized by the hereditary absence of the proximal interphalangeal (PIP) joints (Cushing symphalangism). Severity of PIP joint involvement diminishes towards the radial side. Distal interphalangeal joints are less frequently involved and metacarpophalangeal joints are rarely affected whereas carpal bone malformation and fusion are common. In the lower extremities, tarsal bone coalition is common. Conducive hearing loss is seen and is due to fusion of the stapes to the petrous part of the temporal bone.[7] [8] [9] Defects in GDF5 are the cause of multiple synostoses syndrome type 2 (SYNS2) [MIM:610017. Multiple synostoses syndrome is an autosomal dominant condition characterized by progressive joint fusions of the fingers, wrists, ankles and cervical spine, characteristic facies and progressive conductive deafness.[:][10] Defects in GDF5 are a cause of brachydactyly type A2 (BDA2) [MIM:112600. Brachydactylies (BDs) are a group of inherited malformations characterized by shortening of the digits due to abnormal development of the phalanges and/or the metacarpals. They have been classified on an anatomic and genetic basis into five groups, A to E, including three subgroups (A1 to A3) that usually manifest as autosomal dominant traits.[11] [12] Genetic variations in GDF5 are associated with susceptibility to osteoarthritis type 5 (OS5) [MIM:612400. Osteoarthritis is a degenerative disease of the joints characterized by degradation of the hyaline articular cartilage and remodeling of the subchondral bone with sclerosis. Clinical symptoms include pain and joint stiffness often leading to significant disability and joint replacement. Defects in GDF5 may be a cause of brachydactyly type A1 (BDA1) [MIM:112500. Brachydactylies (BDs) are a group of inherited malformations characterized by shortening of the digits due to abnormal development of the phalanges and/or the metacarpals. They have been classified on an anatomic and genetic basis into five groups, A to E, including three subgroups (A1 to A3) that usually manifest as autosomal dominant traits.[13] FunctionGDF5_HUMAN Could be involved in bone and cartilage formation. Chondrogenic signaling is mediated by the high-affinity receptor BMPR1B.[14] [15] Evolutionary Conservation Check, as determined by ConSurfDB. You may read the explanation of the method and the full data available from ConSurf. Publication Abstract from PubMedDysregulation of growth and differentiation factor 5 (GDF-5) signalling, a member of the TGF-beta superfamily, is strongly linked to skeletal malformation. GDF-5-mediated signal transduction involves both BMP type I receptors, BMPR-IA and BMPR-IB. However, mutations in either GDF-5 or BMPR-IB lead to similar phenotypes, indicating that in chondrogenesis GDF-5 signalling seems to be exclusively mediated through BMPR-IB. Here, we present structural insights into the GDF-5:BMPR-IB complex revealing how binding specificity for BMPR-IB is generated on a molecular level. In BMPR-IB, a loop within the ligand-binding epitope functions similar to a latch allowing high-affinity binding of GDF-5. In BMPR-IA, this latch is in a closed conformation leading to steric repulsion. The new structural data now provide also a molecular basis of how phenotypically relevant missense mutations in GDF-5 might impair receptor binding and activation. Crystal structure analysis reveals a spring-loaded latch as molecular mechanism for GDF-5-type I receptor specificity.,Kotzsch A, Nickel J, Seher A, Sebald W, Muller TD EMBO J. 2009 Apr 8;28(7):937-47. Epub 2009 Feb 19. PMID:19229295[16] From MEDLINE®/PubMed®, a database of the U.S. National Library of Medicine. See AlsoReferences
|
| ||||||||||||||||