User:Nathan Roy/Sandbox 1I am a graduate student at the University of Vermont, studying the dynamics of HIV-1 cell to cell transmission and HIV-1 induced syncytia formation. Our wonderful professor, Dr. Steven Everse, has commissioned us (his students from his BioChem 351 course) to create a page describing the structure of a protein that interests us. I have chosen the HIV-1 gag protein, and more specifically, the MA and CA domains.
HIV-1 Gag
The HIV-1 Gag protein is the major structural protein required for virus assembly. It is synthesized as a polyprotein in the cytosol of an infected cell, and contains four functional segments; MA, CA (NTD and CTD), NC, and p6. The NC region is flanked by two "spacer" segments, denoted SP1 and SP2. The polyprotein is all alpha helical, except the NC region, which is composed of two RNA interacting zinc knuckle domains. Gag is often referred to as an "assembly machine", because expression of Gag alone is sufficient to produce budding virus-like particles (VLP's), due to multimerization of roughly 2000 Gag molecules per virion. Here, we will take a closer look at the MA and CA domains, and how the structural components of these domains aid in the assembly of virus particles.
|
Viral particles can be classified as immature (pre-budding and non-infectious), and mature (post-budding and infectious), and this exchange is mediated by the HIV-1 protease(LINK). Upon viral budding, Gag is cleaved by the HIV-1 protease at multiple sites, thus possibly changing many of the structural interactions that make up the "immature" particle. For simplicity, we will only be discussing the immature formation of Gag on the plasma membrane of infected cells, as it coordinates organized viral budding.
The MA domain (also called Matrix) is essential for proper targeting of Gag (and thus virus release) to distinct locations on the plasma membrane. The MA domain is myristylated post translationally, which is important for a stable association with the plasma membrane. Perhaps just as important for proper virus assembly, is the interaction of MA with phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2). It has long been known that HIV-1 budding occurs at sites rich in PI(4,5)P2 and cholesterol (lipid rafts, and tetraspanin enriched microdomains, TEMs), probably to ensure proper virus budding localized to the plasma membrane (elimination of PI(4,5)P2 causes virions to assembly at intracellular endosomes). More recently, structural analysis of MA has revealed a myristylation switch that allows exposer of the myristyl group to the plasma membrane only upon binding of MA to PI(4,5)P2, a mechanism ensuring proper localization of Gag to rafts within the plasma membrane.
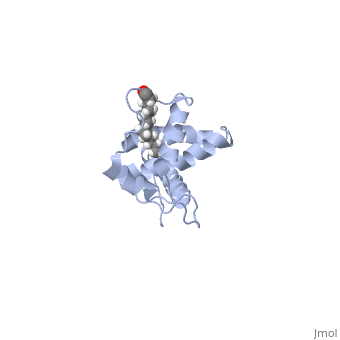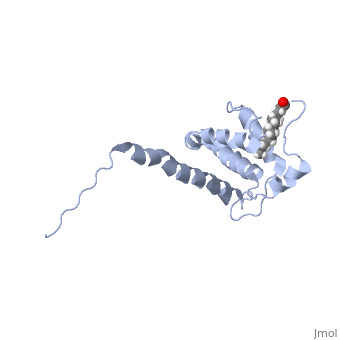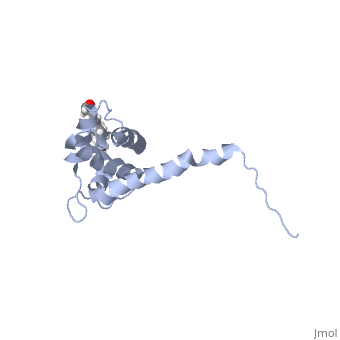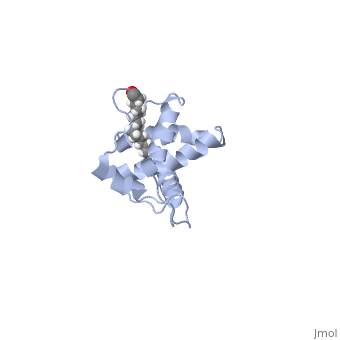
When MA is not bound to PI(4,5)P2 (Figure 1), notice the alignment of helix 1, and more precisely, the orientation of Leu 8 and Glu 12(). In this PI(4,5)P2 unbound structure, the myristyl group is sequestered in the pocket of helix 1 created by Leu 8 and Glu 12. Upon binding of PI(4,5)P2 to the hydrophobic groove created by helix 2, a type 2 beta turn, and helix 5, a slight conformational switch occurs in helix 1 (Figure 2),
|
causing a change in the alignment of Leu 8 and Glu 12,() ejecting the myristyl group from it's sequestered state. This structural switch allows membrane anchoring to be directly coupled to proper membrane localization of Gag, and thus efficient particle release.
Once Gag is localized to discreet sites on the plasma membrane, multimerization of Gag takes place quite quickly , driven by the CA domain, and more specifically our focus here, the C-terminal domain of CA (CTD). There are four helices that contribute to the interaction of CA CTD with it's partner. A side-by-side interaction has been proposed (Figure 3), but many believe the forces involved in the side-by-side model are not great enough to account for the organization and structural stability of assembled Gag. Also, helix 1 of the CA CTD contains a very conserved region of residues within many retroviruses called the MHR (major homology region). In the side-by-side model, the MHR is not responsible for the dimer organization, therefore a model of the CA CTD dimer in which the MHR is responsible for organization of the CA CTD dimers has been sought.
|
By making a single deletion of the Ala 177 residue (which lies in the loop between helix 1 and helix 2), the CA CTD domain adopts a domain-swapped conformation, in which the MHR of helix 1 is extended to contact helices 2,3, and 4 of the adjacent CA CTD domain (Figure 4). This domain swapping allows for a tighter binging of the CA CTD domains, and a stronger, and more rigid viral capsid.
|
|