Prion protein
The prion protein (PrP) is a cell surface glycoprotein. PrP can exist in two alternatively folded confirmations: the cellular isoform (PrPC) can undergo a structural conversion to a 'scrapie' or disease associated isoform termed PrPSc. Prion diseases such as Creutzfeldt Jakob disease (CJD) in people, and bovine spongiform encephalopathy (BSE) commonly known as "mad cow" disease, are characterterized by aggregates of PrPSc, which arise from autocatalytic refolding of PrPC in a template-dependent manner.
Structure of PrPCStructure of PrPC
PrPC has a natively unstructured N-terminal region, and a predominantly α-helical C-terminal region from residues ~120-230.
The N-terminal region can bind coper ions
| |||||||||
| 1hjm, 1 NMR models () | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Related: | 1e1g, 1e1j, 1e1p, 1e1s, 1e1u, 1e1w, 1hjn | ||||||||
| |||||||||
| |||||||||
| Resources: | FirstGlance, OCA, RCSB, PDBsum | ||||||||
| Coordinates: | save as pdb, mmCIF, xml | ||||||||
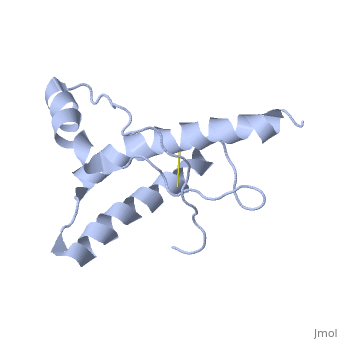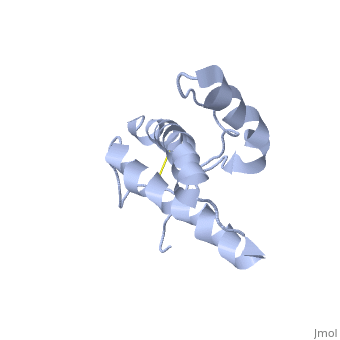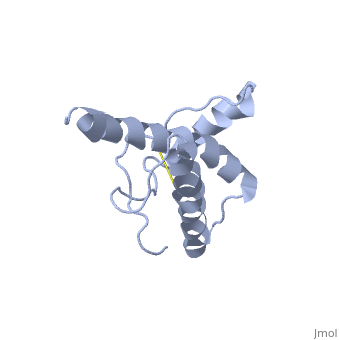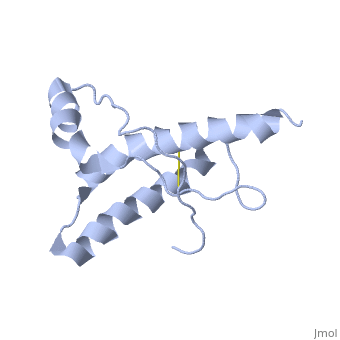

The structure is highly conserved amongst mammals, and only differs slightly in birds, reptiles and amphibians.
The X-ray structure of sheep PrP was dimeric...
Models of PrPSc structureModels of PrPSc structure
Circular dichroism studies first demonstrated that PrPSc had very different proportions of α-helices and β-sheet to PrPC
There are a number of technical obstacles in determining the molecular structure of PrP(sup)Sc
Genetic prion diseasesGenetic prion diseases
A number of mutations in PrP have been identified which correlate with a high incidence of prion disease. To date, structural studies of all mutant PrPC have extremely similar structures to wild type PrPC, suggesting a kinetic basis for the difference in converting to PrPSc.
Prion strainsPrion strains
The strain phenomenon of prions ( ) was initially difficult to equate with the
Selected PrP structuresSelected PrP structures
All structures determined by NMR unless otherwise specified
Human PrPHuman PrP
- 1QLX HuPrP residues 23-230
- 1QM0 HuPrP residues 90-230
- 1QM2 HuPrP residues 121-230
- 1I4M HuPrP residues 119-226 (X-ray)
- 1E1J HuPrP,M166V residues 125-228
- 1E1S HuPrP,S170N residues 125-228
- 1E1W HuPrP,R220K residues 125-228
- 1FKC HuPrP,E200K residues 90-231 (genetic prion disease)
- 1H0L HuPrP residues 121-230, with an additional disulphide bond analogous to the homolog Doppel