User:James D Watson/Structural Templates: Difference between revisions
No edit summary |
|||
| Line 48: | Line 48: | ||
Serine proteases are found in a number of organisms but common to their function is the hydrolysis of peptide bonds. These enzymes catalyse the reaction using a highly reactive serine residue to attack the carbonyl group of the backbone to be hydrolysed. The chemistry of this reaction and the regeneration of the active site, requires the presence of the Ser-His-Asp catalytic triad. In chymotrypsin these residues are (Ser-195, His-57 and Asp-102) whereas in the bacterial subtilisin the site is formed by (Ser-221, His-64 and Asp-32). These two proteins are evolutionary unrelated and this is the classic example of convergent evolution to solve the problem of peptide bond hydrolysis. | Serine proteases are found in a number of organisms but common to their function is the hydrolysis of peptide bonds. These enzymes catalyse the reaction using a highly reactive serine residue to attack the carbonyl group of the backbone to be hydrolysed. The chemistry of this reaction and the regeneration of the active site, requires the presence of the Ser-His-Asp catalytic triad. In chymotrypsin these residues are (Ser-195, His-57 and Asp-102) whereas in the bacterial subtilisin the site is formed by (Ser-221, His-64 and Asp-32). These two proteins are evolutionary unrelated and this is the classic example of convergent evolution to solve the problem of peptide bond hydrolysis. | ||
The detection of these types of motif is almost impossible by looking at the amino acid sequence: there is no evolutionary relationship to detect, the residues are ordered differently in the sequence, and the spacing between the residues also varies. These motifs can be detected relativeley easily | The detection of these types of motif is almost impossible by looking at the amino acid sequence: there is no evolutionary relationship to detect, the residues are ordered differently in the sequence, and the spacing between the residues also varies. These motifs can be detected relativeley easily using structural comparison, particularly the template-based motif detection algorithms (some of which are listed in table 2 below). The subtilisin and chymotrypsin catalytic triads are shown superposed here - note that the global folds of these two proteins are very different so the site could not have been detected using such methods. | ||
Revision as of 01:19, 9 February 2009
Motifs In ProteinsMotifs In Proteins
The term "motif" when used in structural biology tends to refer to one of two cases:
- A particular amino-acid sequence that characterises a biochemical function
- A set of secondary structure elements that defines a functional or structural role
There are a great number of protein sequence motifs identified, many of which have well defined structural or functional roles. One such example of this is the so-called zinc finger motif which is readily identified from the following consensus sequence pattern (where "X" represents any amino acid):
Cys - X(2-4) - Cys - X(3) - Phe - X(5) - Leu - X(2) - His - X(3) - His
|
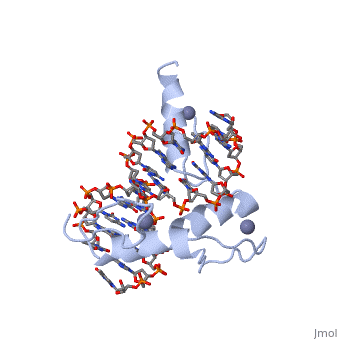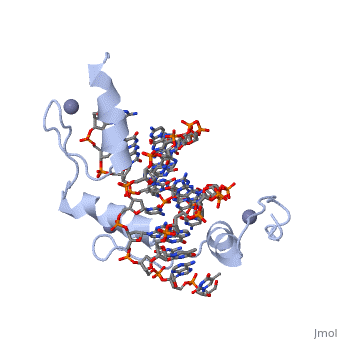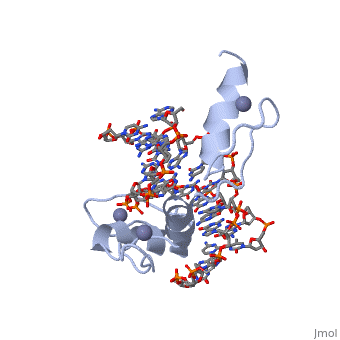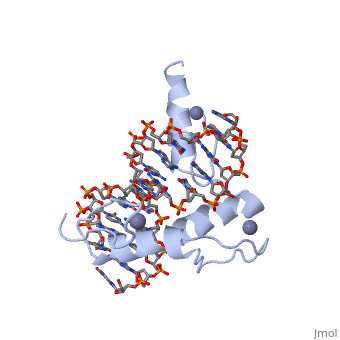
The example structure shown to is that of Zif268 protein-DNA complex from Mus musculus (PDB entry 1AAY). In this example (a C2H2 class zinc finger) the conserved and residues form ligands to a whose coordination is essential to stabilise the tertiary fold of the protein. The fold is important because it helps orientate the to bind to the .
However, there are also a number of repeated patterns and functional motifs revealed when the protein structure is examined. This page aims to introduce some of the main types of motif illustrating them on protein structures from the PDB.
Secondary structure elementsSecondary structure elements
Proteins are formed from linear chains of amino acids joined together by peptide bonds. These chains then fold up to form the three dimensional shape. However, the relative rigidity of the peptide bond combined with the presence of amino acid sidechains, means that not all conformations are acceptable and there are many cases where the various atoms in the chain start to collide with one another. Two of the most stable (and therefore most commonly observed) conformations are the α-helix and the β-pleated sheet. These along with a number of small turns in the chain and random coil (folds that do not fit into a classification) are known as secondary structures.
α-helices The α-helix is formed when the amino acid backbone forms a right handed spiral with 3.6 amino acids per turn. The sidechains point outward, away from the centre of the helix, where athey can interact with solvent, other protein, small molecules or macromolecules. The structure is stabilised by regular hydrogen bonds that form between the backbone carbonyl oxygens and amide hydrogens. The bonding pattern for the α-helix is characterised by the carbonyl group of residue i hydrogen interacting with the amide group of residue i+4, this is known as an (i, i+4) interaction. The alpha-helix can take other less common forms including π-helices, 310-helices and their left handed forms (see table 1 for the helix parameters)
β-sheets
A single beta-strand can be described as a flat helix with 2 residues per turn although this may not be initially obvious. When two or more beta strands lie next to each other, forming hydrogen bonds between them, this is what is termed a β-sheet. As the backbones need to come close together to interact and form a sheet, the sidechains are oriented away from the plane of the sheet. As the polypeptide chain is synthesised from the amino terminus to the carboxyl terminus it has a directionality (represented in cartoon form as an arrowhead on beta strands). β-sheets therefore occur in two varieties:
- Anti-parallel - here the beta strands aligned next to each other run in opposite directions. As the interacting carbonyls and amides align well, the hydrogen bonds appear to be straight.
- Parallel - here the interacting strands run alongside each other and point in the same direction. In this conformation the carbonyl oxygen and the amides tend to be more staggered than in an anti-parallel sheet, therefore the hydrogen bonds tend to be angled.
Turns and loops
There are a number of small hydrogen bonded motifs and patterns which are observed regularly. These are described below:
- Beta Turns - originally defined by the one hydrogen bond common to all (an i, i+3 hydrogen bond) but some modern descriptions do not require a hydrogen bond.
- Beta Bulge Loops - often associated with beta sheets and result from an additional residue being found in one strand. This interrupts the regular hydrogen bonding and causes a distinctive bulge.
- Alpha turns - the simplest of all motifs and is characterised by one (i, i+4) hydrogen bond. It is found as part of the hydrogen bonding network of alpha helices as well as occurring on its own.
- Paperclip/Schellman Motifs - a common motif found at the C-termini of alpha helices which is essentially a reverse turn that breaks the alpha helix out of its cycle. It is characterised by the presence of a left handed residue and two hydrogen bonds: an i, i+3 bond and an i, i+5 bond.
- Gamma Turns - these rarer type of turns are characterised by an (i, i+2) hydrogen bond, which is rather weak because of the bent geometry involved.
These secondary structure motifs can be combined to form functional motifs, the most well known of which is the helix-turn-helix motif found in a number of DNA-binding proteins. The computational identification of these motifs is straightforward but made complicated by the fact that not all helix-turn-helix motifs bind DNA. The problem faced here is therefore one involving the distinguishing between true and false positives.
Templates and Active SitesTemplates and Active Sites
Moving away from large scale secondary structure elements, other structural motifs are more difficult to detect as they are discontinuous and involve elements separated by spacing of various lengths. One such example is that of the "catalytic triad" of the serine proteases.
Serine proteases are found in a number of organisms but common to their function is the hydrolysis of peptide bonds. These enzymes catalyse the reaction using a highly reactive serine residue to attack the carbonyl group of the backbone to be hydrolysed. The chemistry of this reaction and the regeneration of the active site, requires the presence of the Ser-His-Asp catalytic triad. In chymotrypsin these residues are (Ser-195, His-57 and Asp-102) whereas in the bacterial subtilisin the site is formed by (Ser-221, His-64 and Asp-32). These two proteins are evolutionary unrelated and this is the classic example of convergent evolution to solve the problem of peptide bond hydrolysis.
The detection of these types of motif is almost impossible by looking at the amino acid sequence: there is no evolutionary relationship to detect, the residues are ordered differently in the sequence, and the spacing between the residues also varies. These motifs can be detected relativeley easily using structural comparison, particularly the template-based motif detection algorithms (some of which are listed in table 2 below). The subtilisin and chymotrypsin catalytic triads are shown superposed here - note that the global folds of these two proteins are very different so the site could not have been detected using such methods.