Conservation, Evolutionary: Difference between revisions
Eric Martz (talk | contribs) explained the ConSurf process |
Eric Martz (talk | contribs) →The ConSurf Server: polishing |
||
| Line 4: | Line 4: | ||
==The ConSurf Server== | ==The ConSurf Server== | ||
In brief, the [http://consurf.tau.ac.il ConSurf Server] | The [http://consurf.tau.ac.il ConSurf Server] can calculate and display the conservation pattern for 3D structures '''completely automatically'''. It uses state-of-the-art methods, all of which are published in peer-reviewed journal articles. It also permits considerable customization. For example, the user may submit their own multiple sequence alignment, or phylogenetic tree. I<ref>[[User:Eric Martz]] in January, 2009.</ref> know of no other server with these advantages. | ||
In brief, the [http://consurf.tau.ac.il ConSurf Server] uses the following process by default: | |||
# Obtains the protein sequence for the specified PDB code and chain. | # Obtains the protein sequence for the specified PDB code and chain. | ||
# Gathers closely related sequences from Swiss-Prot (or Uniprot) with a PSI-BLAST search. | # Gathers closely related sequences from Swiss-Prot (or Uniprot) with a PSI-BLAST search. | ||
| Line 11: | Line 13: | ||
# Calculates a conservation score for each amino acid. | # Calculates a conservation score for each amino acid. | ||
# Displays the protein in interactive 3D, using [[FirstGlance in Jmol]], [[Chimera]], [[PyMOL]], or [[Protein Explorer]]. | # Displays the protein in interactive 3D, using [[FirstGlance in Jmol]], [[Chimera]], [[PyMOL]], or [[Protein Explorer]]. | ||
==Locating Variable Patches== | ==Locating Variable Patches== | ||
Revision as of 00:44, 1 January 2009
Locating Conserved PatchesLocating Conserved Patches
Patches of highly conserved residues on the surface of a protein molecular structure are good candidates for functional sites. These can be readily identified and visualized automatically with the ConSurf Server, provided a sufficient number of related protein sequences are in the Uniprot database. Before submitting a custom job request to the ConSurf Server, check out your PDB code at ConSurfDB, a database of precalculated conservation levels for most chains in the Protein Data Bank.
The ConSurf ServerThe ConSurf Server
The ConSurf Server can calculate and display the conservation pattern for 3D structures completely automatically. It uses state-of-the-art methods, all of which are published in peer-reviewed journal articles. It also permits considerable customization. For example, the user may submit their own multiple sequence alignment, or phylogenetic tree. I[1] know of no other server with these advantages.
In brief, the ConSurf Server uses the following process by default:
- Obtains the protein sequence for the specified PDB code and chain.
- Gathers closely related sequences from Swiss-Prot (or Uniprot) with a PSI-BLAST search.
- Does a multiple sequence alignment.
- Constructs a phylogenetic tree.
- Calculates a conservation score for each amino acid.
- Displays the protein in interactive 3D, using FirstGlance in Jmol, Chimera, PyMOL, or Protein Explorer.
Locating Variable PatchesLocating Variable Patches
In some cases, patches of highly variable (rapidly mutating) residues are also functional sites. These can also be identified with the ConSurf Server. For example, mutations in influenza hemagglutinin help the virus to evade host defenses. Another example is the high allelic variability of the peptide-binding groove of Major Histocompatibility Complex Class I. That variability helps the grooves of the alleles within any individual to bind a wide range of peptides, hence enabling the T lymphocyte system to defend against a wide range of pathogens, including influenza virus.
ExamplesExamples
|
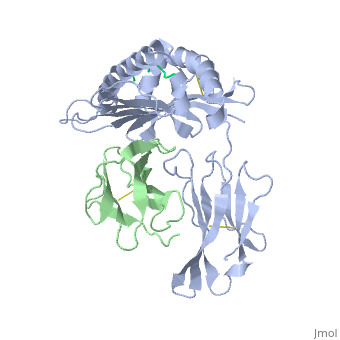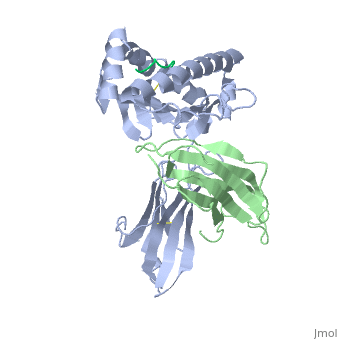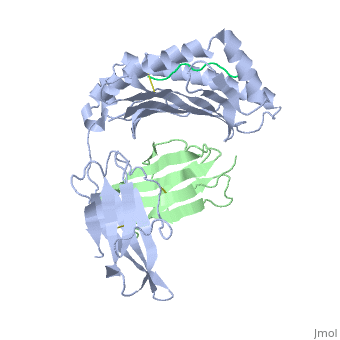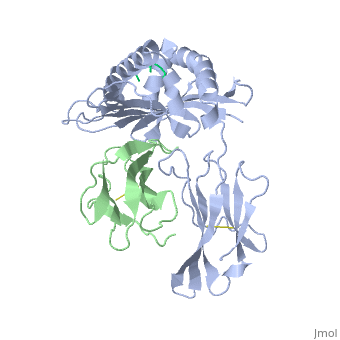
At right is the pattern of evolutionary conservation and variability reported by ConSurfDB for the alpha chain of Major Histocompatibility Complex Class I (chain A of 2vaa). Below are instructions for how to insert a ConSurf result into a Proteopedia scene.
Examples of conserved patches revealed by ConSurf will be found in the articles on
Conservation for Domain FoldingConservation for Domain Folding
Certain residues on the surfaces of protein molecules tend to be conserved in order to maintain proper folding, rather than because they are part of a site functioning to interact with substrate, ligand, or a protein partner. Secondary structure elements need to break, in order to turn back into the folded protein domain, at the protein molecular surface. Therefore, it is common to see highly conserved residues that enable turns, or break helices, notably glycines or prolines, on protein structure surfaces.
Every structure in Proteopedia has a link to be displayed in FirstGlance in Jmol. There, you can use the Find dialog to enter the name of an amino acid, e.g. glycine or proline, and the positions of all of the specified amino acids will be highlighted. You can then visualize their distribution in the 3D structure.
NotesNotes
- ↑ User:Eric Martz in January, 2009.