Hemoglobin: Difference between revisions
Eran Hodis (talk | contribs) |
Eran Hodis (talk | contribs) |
||
| Line 42: | Line 42: | ||
==Troubled Hemoglobins== | ==Troubled Hemoglobins== | ||
[[Image:2hbs-fiber.gif]] | [[Image:2hbs-fiber.gif|right]] | ||
The genes for the protein chains of hemoglobin show small differences within different human populations, so the amino acid sequence of hemoglobin is slightly different from person to person. In most cases the changes do not affect protein function and are often not even noticed. However, in some cases these different amino acids lead to major structural changes. One such example is that of the sickle cell hemoglobin, where glutamate 6 in the beta chain is mutated to valine. This change allows the deoxygenated form of the hemoglobin to stick to each other, as seen in PDB entry [[2hbs]], producing stiff fibers of hemoglobin inside red blood cells. This in turn deforms the red blood cell, which is normally a smooth disk shape, into a C or sickle shape. The distorted cells are fragile and often rupture, leading to loss of hemoglobin. This may seem like a uniformly terrible thing, but in one circumstance, it is actually an advantage. The parasites that cause the tropical disease malaria, which spend part of their life cycle inside red blood cells, cannot live in the fiber-filled sickle cells. Thus people with sickle cell hemoglobin are somewhat resistant to malaria. Other circumstances leading to troubled hemoglobins arise from a mismatch in the production of the alpha and beta proteins. The structure obviously requires equal production of both proteins. However, if one of these proteins is missing, it leads to conditions called Thalassemia. | The genes for the protein chains of hemoglobin show small differences within different human populations, so the amino acid sequence of hemoglobin is slightly different from person to person. In most cases the changes do not affect protein function and are often not even noticed. However, in some cases these different amino acids lead to major structural changes. One such example is that of the sickle cell hemoglobin, where glutamate 6 in the beta chain is mutated to valine. This change allows the deoxygenated form of the hemoglobin to stick to each other, as seen in PDB entry [[2hbs]], producing stiff fibers of hemoglobin inside red blood cells. This in turn deforms the red blood cell, which is normally a smooth disk shape, into a C or sickle shape. The distorted cells are fragile and often rupture, leading to loss of hemoglobin. This may seem like a uniformly terrible thing, but in one circumstance, it is actually an advantage. The parasites that cause the tropical disease malaria, which spend part of their life cycle inside red blood cells, cannot live in the fiber-filled sickle cells. Thus people with sickle cell hemoglobin are somewhat resistant to malaria. Other circumstances leading to troubled hemoglobins arise from a mismatch in the production of the alpha and beta proteins. The structure obviously requires equal production of both proteins. However, if one of these proteins is missing, it leads to conditions called Thalassemia. | ||
==Exploring the Structure== | ==Exploring the Structure== | ||
Revision as of 03:23, 20 November 2007
|
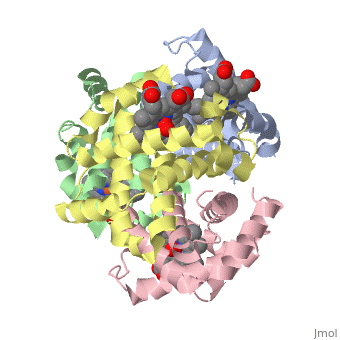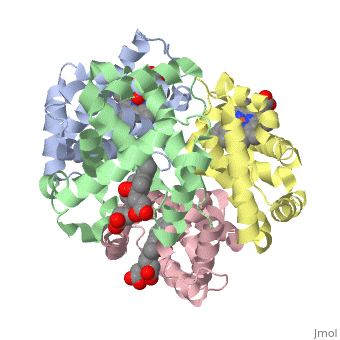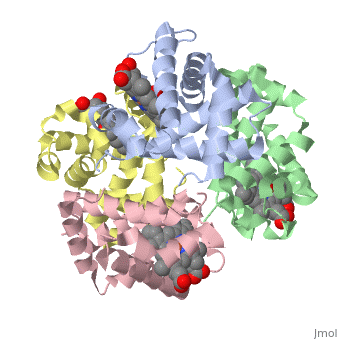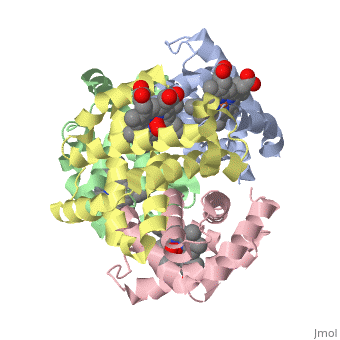
Hemoglobin is an oxygen-transport protein. Hemoglobin is an allosteric protein. It is a tetramer composed of two types of subunits designated α and β, whose stoichiometry is . The of hemoglobin sit roughly at the corners of a tetrahedron, facing each other across a at the center of the molecule. Each of the subunits prosthetic group. The give hemoglobin its red color.
Each individual molecule contains one atom. In the lungs, where oxygen is abundant, an binds to the ferrous iron atom of the heme molecule and is later released in tissues needing oxygen. The heme group binds oxygen while still attached to the . The spacefill view of the hemoglobin polypeptide subunit with an oxygenated heme group shows how the within the polypeptide.
is facilitated by a histidine nitrogen that binds to the iron. A second histidine is near the bound oxygen. The "arms" (propanoate groups) of heme face are hydrophilic and face the surface of the protein while the hydrophobic portions of the heme are buried among the hydrophobic amino acids of the protein. We can also view the anchored heme with the hemoglobin polypeptide subunit shown in a representation.
Secondary structureSecondary structure
Most of the amino acids in hemoglobin form , connected by short non-helical segments. Hemoglobin has no beta strands and no disulfide bonds. A rainbow coloring scheme from N-terminus (blue) to C-terminus (red) helps to discern the . This is at the protein-water interface.
Hydrophobicity, polarity and chargeHydrophobicity, polarity and charge
This view of the shows charged amino acids (lime green), polar amino acids (dark green), hydrophobic amino acids (grey), heme (tan), and water oxygens (light blue). Charged and polar amino acids are common on the surface, where they form hydrogen bonds with water. Most water is not shown. Protein structures tend to fold in such a way so that their hydrophobic residues are buried and their hydrophilic residues are on the surface. A vertical slice through the molecule near the front surface, shows . As we move the slice plane into the molecule, the center is hydrophobic while the surfaces have many polar and charged residues. We can move the slab , and . Near the rear surface, hydrophilic residues are .
Red Blood, Blue BloodRed Blood, Blue Blood
{kind=link}
Ever wondered why blood vessels appear blue? Oxygenated blood is bright red: when you are cut, the blood you see is brilliant red oxygenated blood. Deoxygenated blood is deep purple: when you donate blood or give a blood sample at the doctor's office, it is drawn into a storage tube away from oxygen, so you can see this dark purple color. However, deep purple deoxygenated blood appears blue as it flows through our veins, especially in people with fair skin. This is due to the way that different colors of light travel through skin: blue light is reflected in the surface layers of the skin, whereas red light penetrates more deeply. The dark blood in the vein absorbs most of this red light (as well as any blue light that makes it in that far), so what we see is the blue light that is reflected at the skin's surface. Some organisms like snails and crabs, on the other hand, use copper to transport oxygen, so they truly have blue blood.
Hemoglobin is the protein that makes blood red. It is composed of four protein chains, two alpha chains and two beta chains, each with a ring-like heme group containing an iron atom. Oxygen binds reversibly to these iron atoms and is transported through blood. Each of the protein chains is similar in structure to myoglobin (presented in the January 2000 Molecule of the Month), the protein used to store oxygen in muscles and other tissues. However, the four chains of hemoglobin give it some extra advantages, as described on the next page.
Use and Abuse of HemoglobinUse and Abuse of Hemoglobin
Aside from oxygen transport, hemoglobin can bind and transport other molecules like nitric oxide and carbon monoxide. Nitric oxide affects the walls of blood vessels, causing them to relax. This in turn reduces the blood pressure. Recent studies have shown that nitric oxide can bind to specific cysteine residues in hemoglobin and also to the irons in the heme groups, as shown in PDB entry 1buw. Thus, hemoglobin contributes to the regulation of blood pressure by distributing nitric oxide through blood. Carbon monoxide, on the other hand, is a toxic gas. It readily replaces oxygen at the heme groups, as seen in PDB entry 2hco and many others, forming stable complexes that are difficult to remove. This abuse of the heme groups blocks normal oxygen binding and transport, suffocating the surrounding cells.
Artificial BloodArtificial Blood
Blood transfusions have saved countless lives. However, the need for matching blood type, the short life of stored blood, and the possibility of contamination are still major concerns. An understanding of how hemoglobin works, based on decades of biochemical study and many crystallographic structures, has prompted a search for blood substitutes and artificial blood. The most obvious approach is to use a solution of pure hemoglobin to replace lost blood. The main challenge is keeping the four protein chains of hemoglobin together. In the absence of the protective casing of the red blood cell, the four chains rapidly fall apart. To avoid this problem, novel hemoglobin molecules have been designed where the two of the four chains are physically linked together, as shown in PDB entry 1c7d. In that structure, two additional glycine residues form a link between two of the chains, preventing their separation in solution.
Hemoglobin CousinsHemoglobin Cousins
Looking through the PDB, you will find many different hemoglobin molecules. You can find Max Perutz's groundbreaking structure of horse hemoglobin in entry 2dhb, shown in the picture here. There are structures of human hemoglobins, both adult and fetal. You can also find unusual hemoglobins like leghemoglobin, which is found in legumes. It is thought to protect the oxygen-sensitive bacteria that fix nitrogen in leguminous plant roots. In the past few years some hemoglobin cousins called the "truncated hemoglobins" have been identified, such as the hemoglobin in PDB entry 1idr, which have several portions of the classic structure edited out. The only feature that is absolutely conserved in this subfamily of proteins is the histidine amino acid that binds to the heme iron.
Cooperation Makes It EasierCooperation Makes It Easier
{kind=link}
Hemoglobin is a remarkable molecular machine that uses motion and small structural changes to regulate its action. Oxygen binding at the four heme sites in hemoglobin does not happen simultaneously. Once the first heme binds oxygen, it introduces small changes in the structure of the corresponding protein chain. These changes nudge the neighboring chains into a different shape, making them bind oxygen more easily. Thus, it is difficult to add the first oxygen molecule, but binding the second, third and fourth oxygen molecules gets progressively easier and easier. This provides a great advantage in hemoglobin function. When blood is in the lungs, where oxygen is plentiful, oxygen easily binds to the first subunit and then quickly fills up the remaining ones. Then, as blood circulates through the body, the oxygen level drops while that of carbon dioxide increases. In this environment, hemoglobin releases its bound oxygen. As soon as the first oxygen molecule drops off, the protein starts changing its shape. This promotes the remaining three oxygens to be quickly released. In this way, hemoglobin picks up the largest possible load of oxygen in the lungs, and delivers all of it where and when needed.
In this animated figure, the heme group of one subunit, shown in the little circular window, is kept in one place so that you can see how the protein moves around it when oxygen binds. The oxygen molecule is shown in blue green. As it binds to the iron atom in the center of the heme, it pulls a histidine amino acid upwards on the bottom side of the heme. This shifts the position of an entire alpha helix, shown here in orange below the heme. This motion is propagated throughout the protein chain and on to the other chains, ultimately causing the large rocking motion of the two subunits shown in blue. The two structures shown are PDB entries 2hhb and 1hho.
Troubled HemoglobinsTroubled Hemoglobins
{kind=link}
The genes for the protein chains of hemoglobin show small differences within different human populations, so the amino acid sequence of hemoglobin is slightly different from person to person. In most cases the changes do not affect protein function and are often not even noticed. However, in some cases these different amino acids lead to major structural changes. One such example is that of the sickle cell hemoglobin, where glutamate 6 in the beta chain is mutated to valine. This change allows the deoxygenated form of the hemoglobin to stick to each other, as seen in PDB entry 2hbs, producing stiff fibers of hemoglobin inside red blood cells. This in turn deforms the red blood cell, which is normally a smooth disk shape, into a C or sickle shape. The distorted cells are fragile and often rupture, leading to loss of hemoglobin. This may seem like a uniformly terrible thing, but in one circumstance, it is actually an advantage. The parasites that cause the tropical disease malaria, which spend part of their life cycle inside red blood cells, cannot live in the fiber-filled sickle cells. Thus people with sickle cell hemoglobin are somewhat resistant to malaria. Other circumstances leading to troubled hemoglobins arise from a mismatch in the production of the alpha and beta proteins. The structure obviously requires equal production of both proteins. However, if one of these proteins is missing, it leads to conditions called Thalassemia.
Exploring the StructureExploring the Structure
You can look at the binding of oxygen up close in two structures of human hemoglobin. PDB entry 2hhb shows hemoglobin with no oxygen bound. In this picture, the heme is seen edge-on with the iron atom colored in gold. You can see the key histidine reaching up on the bottom side to bind to the iron atom. In PDB entry 1hho, oxygen has bound to the iron, pulling it upwards. This in turn, pulls on the histidine below, which then shifts the location of the entire protein chain. These changes are transmitted throughout the protein, ultimately causing the big shift in shape that changes the binding strength of the neighboring sites.
This picture was created with Rasmol. You can create similar pictures by clicking on the accession codes and choosing one of the options under View Structure. Note that the PDB entry 1hho only contains two of the four chains in the hemoglobin structure. To get the full tetramer, click on the accession code, and then go to Other Sources. There, you will find a link to the EBI MSD Macromolecule File Server, where you can download a full set of coordinates.
BELOW Sections Under ConstructionBELOW Sections Under Construction
Hemoglobin subunit binding O2Hemoglobin subunit binding O2
Human hemoglobin alpha1 subunit, deoxy (blue shades) vs oxy (pink shades) structures. Animate to switch between the two forms. View1 shows the overall alpha subunit, View2 is a closeup of the heme site, View3 shows the hydrophobic heme pocket, View4 moves out from the heme toward the interface with alpha 2 (alpha 2 not shown), and View5 goes to standard orientation for the tetramer, as in View1 of Kin.13. (from PDB files 3HHB, 1HCO)
|
For hemoglobin, its function as an oxygen-carrier in the blood is fundamentally linked to the equilibrium between the two main states of its quaternary structure, the unliganded "deoxy" or "T state" versus the liganded "oxy" or "R state". The is shown in shades of blue (bluetint alpha-chains, cyan betas, and skyblue hemes) and the is in shades of pink (pinktint alphas, pink betas, and hotpink hemes), suggestive of the change in color between deoxygenated and oxygenated blood. The unliganded (deoxy) form is called the "T" (for "tense") state because it contains extra stabilizing interactions between the subunits. In the high-affinity R-state conformation the interactions which oppose oxygen binding and stabilize the tetramer are somewhat weaker or "relaxed". In some organisms this difference is so pronounced that their Hb molecules dissociate into dimers in the oxygenated form. Animation of the structural changes that occur during this transition can illuminate how such changes result in important functional properties, such as cooperativity of oxygen binding and allosteric control by pH and anions. Hemoglobin is definitely not a pure two-state system, but the T to R transition provides the major, first-level explanation of its function.
The hemoglobin molecule (or "Hb") is a tetramer of two alpha and two beta chains, of 141 and 146 residues in human. They are different but homologous, with a "globin fold" structure similar to myoglobin (Kin. 3.6). The two crystal structures used here are human deoxy hemoglobin (PDB file 3HHB), which is in the T-state quaternary structure with no ligands at the O2 binding sites, and human carbonmonoxy hemoglobin (PDB file 1HCO), which is in the R-state quaternary structure and has ligands at all 4 sites.
Kinemage 1 shows a single alpha chain of hemoglobin, starting with an overview of the subunit. The 6 major and 2 short alpha-helices that make up the structure of a Hb subunit (the "globin fold") are labeled A through H, which is the traditional naming scheme. For example, the proximal histidine (the tightest protein Fe ligand) is often called His F9, since it is residue 9 on helix F (it is residue 87 in the human alpha chain). The helices form an approximately-cylindrical bundle, with the heme and its central Fe atom bound in a hydrophobic pocket between the E and F helices. Turn on "highlights" and click the "animate" button, or press the "a" key on the keyboard, to cycle between the deoxy and oxy forms in this overview.
Click here: *{Kinemage 1, View 2, m= {highlights} on}* for a closeup around the heme O2-binding site. Animate back & forth between the deoxy and oxy forms. For this kinemage the oxy and deoxy alpha1 heme groups were superimposed on each other, to give a local comparison at this site. The heme is quite domed in the blue T-state (deoxy) form, with the 5-coordinate, high-spin Fe (yellow ball) out of the plane. In the pink R-state form a CO molecule is bound at the left; the Fe, now 6-coordinate low-spin, has moved into the heme plane, which has flattenened. The proximal His (at right) connects the Fe to helices on the proximal side, making the Fe position sensitive to changes in the globin structure and vice versa. Remember that this kinemage shows a subunit in the all-unliganded versus the all-liganded states of Hb; when oxygen binds to just one subunit, then its internal structure undergoes some but not all of these changes, depending on conditions.
O2 binds in the same place as CO, with similar effects on the structure; however, for O2 the outer atom is angled rather than straight. The equilibrium between free and bound O2 is very rapid, with on and off rates that are sensitive to protein conformation. Both CO and NO dissociate from the Fe atom very slowly, so that these gases act as respiratory poisons. The alpha and beta chains differ somewhat in their rates and relative affinities for O2 and other ligands, by virtue of heme-pocket differences, but the differences between affinities in the R vs T quaternary states are much larger.
Both alpha and beta chains of Hb resemble myoglobin (the single-chain O2-binder in muscle), both in overall tertiary structure and in using an Fe atom centered in a heme group as the site where oxygen is reversibly bound. The heme is surrounded by a hydrophobic pocket, which is necessary in order for it to bind oxygen reversibly without undergoing oxidation or other undesirable reactions. Click here: *{Kin 1, View 3, m= {Hphobics} on}* to see some of the hydrophobic sidechains that form the heme pocket. They actually surround the binding site so thoroughly that O2 cannot get in or out without parts of the protein moving out of the way a bit, so that its dynamic properties are essential to have any O2 binding at all; this restrictive process also increases the specificity of ligand binding.
The shift between R and T state requires subunit interactions and does not occur in myoglobin, or in isolated alpha or beta chain monomers. These monomers bind O2 quite tightly, which would work well for loading O2 in the lungs but would not allow unloading it for delivery to the tissues. Therefore, the central critical feature of hemoglobin function is how it achieves, uses, and allosterically controls cooperativity between the 4 binding sites in the tetramer to tune O2 binding for satisfying physiological needs.
Click here: *{Kin 1, View 4, m= {Hphobics} off}* and animate, to see the linkage between changes at the O2-binding site and changes in protein conformation, which shows ligand-dependent shifts in the region from the heme out to the subunit interface. Linkage of the heme Fe through the proximal His results in tertiary-structure changes that can then transmit their effects to other subunits in the tetrameric assemblage. This allows O2 binding in one subunit to indirectly affect the affinitiy of other subunits. Briefly, inside the alpha chains the R/T equilibrium is reflected in changes in Fe spin state and position as it moves in or out of the heme plane; the proximal His changes distance and angle relative to the heme; the F helix shifts; Tyr 140 moves and its H-bond to backbone weakens; and both the C-terminus of the chain and Arg 141 move significantly at the interface. Changes at the subunit interface (coupled with changes at the Fe, as we have seen) alter the equilibrium between the deoxy and oxy quaternary structures, and conversely a change of quaternary structure alters the balance between the two states inside a given subunit. Each O2 that binds increases the likelihood of switching the tetramer into the oxy state, and once it switches, the O2 affinity at all sites increases because the local structure changes have either already occurred or are easier to make.
Click here: *{Kin 1, View 5, m= {highlights} off, m= {axes} on}* to show the alpha1 subunit, but centered for the whole tetramer (deoxy form), as it will be seen in View1 of Kinemage 13, including the 2-fold axes of symmetry of the tetramer.
The Hb tetramer T -> R transitionThe Hb tetramer T -> R transition
Kinemage 2 - Hemoglobin tetramer - deoxy (blue shades) vs oxy (pink shades) animation. For a guided tour, use the hypertext feature in the text window: first looking down the central cavity, which is wider in the deoxy state, forming phosphate sites; quaternary structure change as rigid rotations of Alpha-Beta dimers; Alpha1-Beta2 contact overview; "ratchet" vs "hinge" at the a1b2 interface; Alpha1-Alpha2 salt bridges; charged groups at the C-terminus of Beta2 which stabilize the deoxy form; and finally a summary overview. (from PDB files bio3HHB and bio1HCO)
Kinemage 2 shows multiple views of the quaternary-structure change for a tetramer of human hemoglobin. Animate to change between the T-state (deoxy) and R-state (oxy) forms. The starting view looks down one of the approximate 2-fold axes, with alpha subunits at the top and beta subunits at the bottom. Notice that the hemes are quite far apart, so that their interactions must be mediated by the protein. For a view down the exact crystallographic 2-fold axis from the Beta1-Beta2 end, click here: *{Kinemage 2, View 2}*. The yellowtint crosses are phosphate sites present in deoxy but not oxy Hb. In oxy Hb, the beta subunits move closer together, squeezing out phosphates (such as 2,3 DPG), and allowing the N- and C-termini to interact. DPG and other phosphates bind very much more strongly to the deoxy quaternary structure; therefore they necessarily push the equilibrium toward deoxy Hb, and because of that they decrease O2 affinity. Such regulatory phosphate molecules are useful in the blood, because their concentrations can be controlled to shift the Hb O2-binding curve so that it is working across the steepest and most efficient part under conditions in the lungs and tissues. For instance, at high altitude the body makes more DPG, to unload O2 more effectively in the muscles.
Like the PFK of Kin. 11, to the first approximation the Hb molecule consists of two "dimers" (Alpha1-Beta1 and Alpha2-Beta2), which rotate relative to each other as rigid bodies in the R-T transition. To see that rotation for just one dimer, click here: *{Kin 2, View 3, m= {alpha1} off, m= {beta1} off}*. The Alpha1-Beta1 unit undergoes relatively little internal rearrangement, but its overall rotation is considerable. The net rotation of the two dimers alters their interactions with one another, most notably at the allosteric effector site between Beta1 and Beta2 (PO4 binding) and at the important Alpha1-Beta2 interface, where mutations have the largest effect on Hb allosteric properties. For an overview of relative shifts at the critical Alpha1-Beta2 interface, click here: *{Kin 2, View 4, m= {alpha1} on, m= {beta2} on, m= {alpha2} off, m= {beta1} off, m= {salt links} on, m= {hinge&ratchet} on}*. Although the symmetry is not exact, similar parts of the subunits contact each other: the C helix, and the "FG corner" between helices F and G. Animate, to see the relative motion of these two subunits, with a fairly stationary "hinge" near the top and a larger "ratchet" motion near the bottom.
Click here: *{Kin 2, View 5, m= {hinge&ratchet} on, m= {salt links} off, m= {axes} off}* for a closeup that emphasizes the ratchet contact between the C helix of Alpha1 and the FG corner of Beta2; His 97 of the Beta2 FG corner makes a large jump against Thr 38 and Thr 41 of the Alpha1 C helix. Animate repeatedly, keeping your eye on His 97. Click here: *{Kin 2, View 6, m= {hinge&ratchet} on, m= {salt links} off}* for a closeup of the hinge contact, where the motions are mainly rotations without much shift, between the Alpha1 FG corner and the Beta2 C helix. Labels help identify these parts. Since this is a complex motion orchestrated between the fit of two quite different sets of contacts in the two states, this interface is critical to making Hb allostery work, and mutations of residues in this interface have been found to be especially likely to influence cooperativity and allostery.
There are salt links between Alpha1 and Alpha2, which stabilize the deoxy form. Click here: *{Kin 2, View 7, m= {salt links} on, master= {a1a2 salt2} on, m= {hinge&ratchet} off, m= {alpha2} on, master= {beta1} off, master= {beta2} off}* for an overview down the exact 2-fold axis between the subunits, showing that there are two equivalent sets of interactions, on either side of the twofold. Animate, to see the making and breaking of these interactions.
Salt links at the C-terminus of Beta2 stabilize the deoxy T form and make a large contribution to the pH dependence of Hb oxygen binding, known as the Bohr Effect. Click here: *{Kin 2, View 8, m= {salt links} on, m= {a1a2 salt2} off, m= {beta2} on, m= {axes} off}* for a closeup to see the making and breaking of these interactions. His b 146 moves a great deal, disrupting the salt link (charged H-bond) to Asp b 94 that is formed in the T state. Since His titrates near physiological pH, this interaction is quite pH sensitive. At low pH, when more protons are present, the His ring N is more likely to be protonated and positive; this strengthens its H-bond with Asp 94, thus favoring the T state and decreasing O2 affinity. The pH effect, or Bohr Effect, can be considered as allosteric regulation by the binding of protons. It is important biologically, because it promotes oxygen unloading in the tissues where proton concentrations are elevated, for instance by the production of lactic acid in muscle.
To summarize some of these critical subunit interactions in the context of the whole hemoglobin tetramer, click here: *{Kinemage 2, View 9, m= {salt links} on, m= {hinge&ratchet} on, m= {beta1} on, m= {axes} on}*. Animate, to follow components of the T-R changes in the contacts at the allosteric interface between the two dimers.
Kinemages 1 and 2 were adapted from "THE PROTEIN TOURIST #8 - THE T-R, DEOXY-OXY TRANSITION IN HUMAN HEMOGLOBIN", David Richardson, Celia Bonaventura, and Jane Richardson, Protein Science vol. 3, electronic supplement, Oct. 1994.
References, for further information on HemoglobinReferences, for further information on Hemoglobin
To the structures used here:
- Baldwin (1980) "The crystal structure of human carbonmonoxy haemoglobin at 2.7A resolution", J. Mol. Biol. 136: 103. (file 1HCO)
- Fermi, Perutz, Shaanan, & Fourme (1984) "The crystal structure of human deoxy haemoglobin at 1.74A resolution", J. Mol. Biol. 175: 159. (file 3HHB)
General treatments of Hb allostery:
- Perutz (1970) "Stereochemistry of cooperative effects in haemoglobin", Nature 228: 726
- Baldwin & Chothia (1979) "Haemoglobin. The structural changes related to ligand binding and its allosteric mechanism", J. Mol. Biol. 129: 175.
- Dickerson & Geis (1983) "Hemoglobin: Structure, Function, and Pathology", Benjamin/Cummings Publ., Menlo Park, CA
- Perutz (1989) "Mechanisms of cooperativity and allosteric regulation in proteins", Quarterly Rev. of Biophys. 22: 139-236
- Ackers, Doyle, Myers, & Daugherty (1992) "Molecular code for cooperativity in hemoglobin", Science 255: 54
- Perutz, Fermi, Poyart, Pagnier, & Kister (1993) "A novel allosteric mechanism in haemoglobin: Structure of bovine deoxyhaemoglobin, absence of specific chloride binding sites, and origin of the chloride-linked Bohr Effect in bovine and human haemoglobin", J. Mol. Biol. 233: 536
Hb structures in other quaternary states or intermediates:
- Silva, Rogers, & Arnone (1992) "A third quaternary structure of human hemoglobin A at 1.7A resolution", J. Biol. Chem. 267: 17248
- Smith, Lattman, & Carter (1991) "The mutation beta99 Asp-Tyr stabilizes Y - A new, composite quaternary state of human hemoglobin", Proteins: Struct., Funct., Genet. 10: 81
- Liddington, Derewenda, Dodson, Hubbard, & Dodson (1992) "High resolution crystal structures and comparisons of T state deoxyhaemoglobin and two liganded T-state haemoglobins: T(alpha-oxy)haemoglobin and T(met)Haemoglobin", J. Mol. Biol. 228: 551
Copyright 1999 Jane S. and David C. Richardson. Permission freely granted
AcknowledgementsAcknowledgements
- Content adopted from David S. Goodsell and Shuchismita Dutta's Molecule of the Month on Hemoglobin http://mgl.scripps.edu/people/goodsell/pdb/pdb41/pdb41_1.html
- Content adopted from Jane S. and David C. Richardson's http://kinemage.biochem.duke.edu/
- Content adopted from Eric Martz's http://www.umass.edu/molvis/tutorials/hemoglobin/