1iyt: Difference between revisions
m Protected "1iyt" [edit=sysop:move=sysop] |
No edit summary |
||
| (9 intermediate revisions by the same user not shown) | |||
| Line 1: | Line 1: | ||
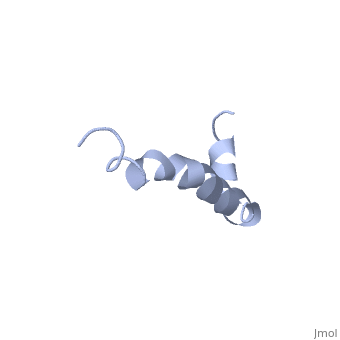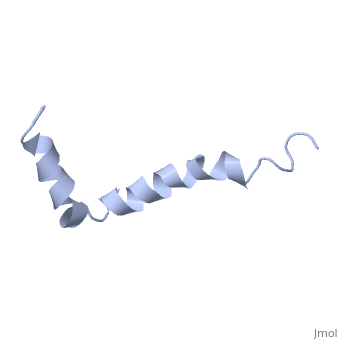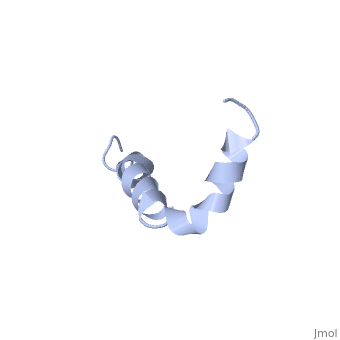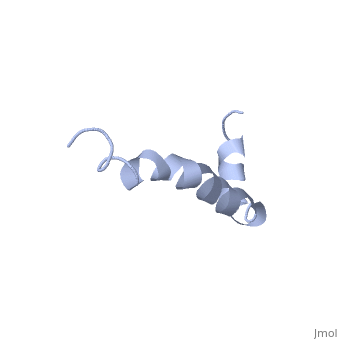
==Solution structure of the Alzheimer's disease amyloid beta-peptide (1-42)== | |||
<StructureSection load='1iyt' size='340' side='right'caption='[[1iyt]]' scene=''> | |||
== Structural highlights == | |||
<table><tr><td colspan='2'>[[1iyt]] is a 1 chain structure with sequence from [https://en.wikipedia.org/wiki/Homo_sapiens Homo sapiens]. The July 2006 RCSB PDB [https://pdb.rcsb.org/pdb/static.do?p=education_discussion/molecule_of_the_month/index.html Molecule of the Month] feature on ''Amyloid-beta Precursor Protein'' by David S. Goodsell is [https://dx.doi.org/10.2210/rcsb_pdb/mom_2006_7 10.2210/rcsb_pdb/mom_2006_7]. Full experimental information is available from [http://oca.weizmann.ac.il/oca-bin/ocashort?id=1IYT OCA]. For a <b>guided tour on the structure components</b> use [https://proteopedia.org/fgij/fg.htm?mol=1IYT FirstGlance]. <br> | |||
</td></tr><tr id='method'><td class="sblockLbl"><b>[[Empirical_models|Method:]]</b></td><td class="sblockDat" id="methodDat">Solution NMR</td></tr> | |||
<tr id='resources'><td class="sblockLbl"><b>Resources:</b></td><td class="sblockDat"><span class='plainlinks'>[https://proteopedia.org/fgij/fg.htm?mol=1iyt FirstGlance], [http://oca.weizmann.ac.il/oca-bin/ocaids?id=1iyt OCA], [https://pdbe.org/1iyt PDBe], [https://www.rcsb.org/pdb/explore.do?structureId=1iyt RCSB], [https://www.ebi.ac.uk/pdbsum/1iyt PDBsum], [https://prosat.h-its.org/prosat/prosatexe?pdbcode=1iyt ProSAT]</span></td></tr> | |||
</table> | |||
== Disease == | |||
[https://www.uniprot.org/uniprot/A4_HUMAN A4_HUMAN] Defects in APP are the cause of Alzheimer disease type 1 (AD1) [MIM:[https://omim.org/entry/104300 104300]. AD1 is a familial early-onset form of Alzheimer disease. It can be associated with cerebral amyloid angiopathy. Alzheimer disease is a neurodegenerative disorder characterized by progressive dementia, loss of cognitive abilities, and deposition of fibrillar amyloid proteins as intraneuronal neurofibrillary tangles, extracellular amyloid plaques and vascular amyloid deposits. The major constituent of these plaques is the neurotoxic amyloid-beta-APP 40-42 peptide (s), derived proteolytically from the transmembrane precursor protein APP by sequential secretase processing. The cytotoxic C-terminal fragments (CTFs) and the caspase-cleaved products such as C31 derived from APP, are also implicated in neuronal death.<ref>PMID:8476439</ref> <ref>PMID:15201367</ref> <ref>PMID:1671712</ref> <ref>PMID:1908231</ref> <ref>PMID:1678058</ref> <ref>PMID:1944558</ref> <ref>PMID:1925564</ref> <ref>PMID:1415269</ref> <ref>PMID:1303239</ref> <ref>PMID:1302033</ref> <ref>PMID:1303275</ref> <ref>PMID:8267572</ref> <ref>PMID:8290042</ref> <ref>PMID:8577393</ref> <ref>PMID:9328472</ref> <ref>PMID:9754958</ref> <ref>PMID:10097173</ref> <ref>PMID:10631141</ref> <ref>PMID:10665499</ref> <ref>PMID:10867787</ref> <ref>PMID:11063718</ref> <ref>PMID:11311152</ref> <ref>PMID:11528419</ref> <ref>PMID:12034808</ref> <ref>PMID:15365148</ref> <ref>PMID:15668448</ref> Defects in APP are the cause of cerebral amyloid angiopathy APP-related (CAA-APP) [MIM:[https://omim.org/entry/605714 605714]. A hereditary localized amyloidosis due to amyloid-beta A4 peptide(s) deposition in the cerebral vessels. The principal clinical characteristics are recurrent cerebral and cerebellar hemorrhages, recurrent strokes, cerebral ischemia, cerebral infarction, and progressive mental deterioration. Patients develop cerebral hemorrhage because of the severe cerebral amyloid angiopathy. Parenchymal amyloid deposits are rare and largely in the form of pre-amyloid lesions or diffuse plaque-like structures. They are Congo red negative and lack the dense amyloid cores commonly present in Alzheimer disease. Some affected individuals manifest progressive aphasic dementia, leukoencephalopathy, and occipital calcifications.<ref>PMID:10821838</ref> <ref>PMID:2111584</ref> <ref>PMID:11409420</ref> <ref>PMID:12654973</ref> <ref>PMID:16178030</ref> | |||
== Function == | |||
[https://www.uniprot.org/uniprot/A4_HUMAN A4_HUMAN] Functions as a cell surface receptor and performs physiological functions on the surface of neurons relevant to neurite growth, neuronal adhesion and axonogenesis. Involved in cell mobility and transcription regulation through protein-protein interactions. Can promote transcription activation through binding to APBB1-KAT5 and inhibits Notch signaling through interaction with Numb. Couples to apoptosis-inducing pathways such as those mediated by G(O) and JIP. Inhibits G(o) alpha ATPase activity (By similarity). Acts as a kinesin I membrane receptor, mediating the axonal transport of beta-secretase and presenilin 1. Involved in copper homeostasis/oxidative stress through copper ion reduction. In vitro, copper-metallated APP induces neuronal death directly or is potentiated through Cu(2+)-mediated low-density lipoprotein oxidation. Can regulate neurite outgrowth through binding to components of the extracellular matrix such as heparin and collagen I and IV. The splice isoforms that contain the BPTI domain possess protease inhibitor activity. Induces a AGER-dependent pathway that involves activation of p38 MAPK, resulting in internalization of amyloid-beta peptide and leading to mitochondrial dysfunction in cultured cortical neurons. Provides Cu(2+) ions for GPC1 which are required for release of nitric oxide (NO) and subsequent degradation of the heparan sulfate chains on GPC1.<ref>PMID:9168929</ref> <ref>PMID:11544248</ref> <ref>PMID:11943163</ref> <ref>PMID:19225519</ref> <ref>PMID:19901339</ref> Beta-amyloid peptides are lipophilic metal chelators with metal-reducing activity. Bind transient metals such as copper, zinc and iron. In vitro, can reduce Cu(2+) and Fe(3+) to Cu(+) and Fe(2+), respectively. Beta-amyloid 42 is a more effective reductant than beta-amyloid 40. Beta-amyloid peptides bind to lipoproteins and apolipoproteins E and J in the CSF and to HDL particles in plasma, inhibiting metal-catalyzed oxidation of lipoproteins. Beta-APP42 may activate mononuclear phagocytes in the brain and elicit inflammatory responses. Promotes both tau aggregation and TPK II-mediated phosphorylation. Interaction with Also bind GPC1 in lipid rafts.<ref>PMID:9168929</ref> <ref>PMID:11544248</ref> <ref>PMID:11943163</ref> <ref>PMID:19225519</ref> <ref>PMID:19901339</ref> Appicans elicit adhesion of neural cells to the extracellular matrix and may regulate neurite outgrowth in the brain (By similarity).<ref>PMID:9168929</ref> <ref>PMID:11544248</ref> <ref>PMID:11943163</ref> <ref>PMID:19225519</ref> <ref>PMID:19901339</ref> The gamma-CTF peptides as well as the caspase-cleaved peptides, including C31, are potent enhancers of neuronal apoptosis.<ref>PMID:9168929</ref> <ref>PMID:11544248</ref> <ref>PMID:11943163</ref> <ref>PMID:19225519</ref> <ref>PMID:19901339</ref> N-APP binds TNFRSF21 triggering caspase activation and degeneration of both neuronal cell bodies (via caspase-3) and axons (via caspase-6).<ref>PMID:9168929</ref> <ref>PMID:11544248</ref> <ref>PMID:11943163</ref> <ref>PMID:19225519</ref> <ref>PMID:19901339</ref> | |||
== Evolutionary Conservation == | |||
[[Image:Consurf_key_small.gif|200px|right]] | |||
Check<jmol> | |||
<jmolCheckbox> | |||
<scriptWhenChecked>; select protein; define ~consurf_to_do selected; consurf_initial_scene = true; script "/wiki/ConSurf/iy/1iyt_consurf.spt"</scriptWhenChecked> | |||
<scriptWhenUnchecked>script /wiki/extensions/Proteopedia/spt/initialview01.spt</scriptWhenUnchecked> | |||
<text>to colour the structure by Evolutionary Conservation</text> | |||
</jmolCheckbox> | |||
</jmol>, as determined by [http://consurfdb.tau.ac.il/ ConSurfDB]. You may read the [[Conservation%2C_Evolutionary|explanation]] of the method and the full data available from [http://bental.tau.ac.il/new_ConSurfDB/main_output.php?pdb_ID=1iyt ConSurf]. | |||
<div style="clear:both"></div> | |||
<div style="background-color:#fffaf0;"> | |||
== Publication Abstract from PubMed == | |||
The major components of neuritic plaques found in Alzheimer disease (AD) are peptides known as amyloid beta-peptides (Abeta), which derive from the proteolitic cleavage of the amyloid precursor proteins. In vitro Abeta may undergo a conformational transition from a soluble form to aggregated, fibrillary beta-sheet structures, which seem to be neurotoxic. Alternatively, it has been suggested that an alpha-helical form can be involved in a process of membrane poration, which would then trigger cellular death. Conformational studies on these peptides in aqueous solution are complicated by their tendency to aggregate, and only recently NMR structures of Abeta-(1-40) and Abeta-(1-42) have been determined in aqueous trifluoroethanol or in SDS micelles. All these studies hint to the presence of two helical regions, connected through a flexible kink, but it proved difficult to determine the length and position of the helical stretches with accuracy and, most of all, to ascertain whether the kink region has a preferred conformation. In the search for a medium which could allow a more accurate structure determination, we performed an exhaustive solvent scan that showed a high propensity of Abeta-(1-42) to adopt helical conformations in aqueous solutions of fluorinated alcohols. The 3D NMR structure of Abeta-(1-42) shows two helical regions encompassing residues 8-25 and 28-38, connected by a regular type I beta-turn. The surprising similarity of this structure, as well as the sequence of the C-terminal moiety, with those of the fusion domain of influenza hemagglutinin suggests a direct mechanism of neurotoxicity. | |||
Solution structure of the Alzheimer amyloid beta-peptide (1-42) in an apolar microenvironment. Similarity with a virus fusion domain.,Crescenzi O, Tomaselli S, Guerrini R, Salvadori S, D'Ursi AM, Temussi PA, Picone D Eur J Biochem. 2002 Nov;269(22):5642-8. PMID:12423364<ref>PMID:12423364</ref> | |||
From MEDLINE®/PubMed®, a database of the U.S. National Library of Medicine.<br> | |||
</div> | |||
<div class="pdbe-citations 1iyt" style="background-color:#fffaf0;"></div> | |||
== References == | |||
<references/> | |||
== | __TOC__ | ||
</StructureSection> | |||
== | |||
< | |||
[[Category: Amyloid-beta Precursor Protein]] | [[Category: Amyloid-beta Precursor Protein]] | ||
[[Category: Homo sapiens]] | |||
[[Category: Large Structures]] | |||
[[Category: RCSB PDB Molecule of the Month]] | [[Category: RCSB PDB Molecule of the Month]] | ||
[[Category: Crescenzi | [[Category: Crescenzi O]] | ||
[[Category: Guerrini | [[Category: D'Ursi AM]] | ||
[[Category: Picone | [[Category: Guerrini R]] | ||
[[Category: Salvadori | [[Category: Picone D]] | ||
[[Category: Temussi | [[Category: Salvadori S]] | ||
[[Category: Tomaselli | [[Category: Temussi PA]] | ||
[[Category: Tomaselli S]] | |||
Latest revision as of 02:39, 28 December 2023
Solution structure of the Alzheimer's disease amyloid beta-peptide (1-42)Solution structure of the Alzheimer's disease amyloid beta-peptide (1-42)
Structural highlights
DiseaseA4_HUMAN Defects in APP are the cause of Alzheimer disease type 1 (AD1) [MIM:104300. AD1 is a familial early-onset form of Alzheimer disease. It can be associated with cerebral amyloid angiopathy. Alzheimer disease is a neurodegenerative disorder characterized by progressive dementia, loss of cognitive abilities, and deposition of fibrillar amyloid proteins as intraneuronal neurofibrillary tangles, extracellular amyloid plaques and vascular amyloid deposits. The major constituent of these plaques is the neurotoxic amyloid-beta-APP 40-42 peptide (s), derived proteolytically from the transmembrane precursor protein APP by sequential secretase processing. The cytotoxic C-terminal fragments (CTFs) and the caspase-cleaved products such as C31 derived from APP, are also implicated in neuronal death.[1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] [16] [17] [18] [19] [20] [21] [22] [23] [24] [25] [26] Defects in APP are the cause of cerebral amyloid angiopathy APP-related (CAA-APP) [MIM:605714. A hereditary localized amyloidosis due to amyloid-beta A4 peptide(s) deposition in the cerebral vessels. The principal clinical characteristics are recurrent cerebral and cerebellar hemorrhages, recurrent strokes, cerebral ischemia, cerebral infarction, and progressive mental deterioration. Patients develop cerebral hemorrhage because of the severe cerebral amyloid angiopathy. Parenchymal amyloid deposits are rare and largely in the form of pre-amyloid lesions or diffuse plaque-like structures. They are Congo red negative and lack the dense amyloid cores commonly present in Alzheimer disease. Some affected individuals manifest progressive aphasic dementia, leukoencephalopathy, and occipital calcifications.[27] [28] [29] [30] [31] FunctionA4_HUMAN Functions as a cell surface receptor and performs physiological functions on the surface of neurons relevant to neurite growth, neuronal adhesion and axonogenesis. Involved in cell mobility and transcription regulation through protein-protein interactions. Can promote transcription activation through binding to APBB1-KAT5 and inhibits Notch signaling through interaction with Numb. Couples to apoptosis-inducing pathways such as those mediated by G(O) and JIP. Inhibits G(o) alpha ATPase activity (By similarity). Acts as a kinesin I membrane receptor, mediating the axonal transport of beta-secretase and presenilin 1. Involved in copper homeostasis/oxidative stress through copper ion reduction. In vitro, copper-metallated APP induces neuronal death directly or is potentiated through Cu(2+)-mediated low-density lipoprotein oxidation. Can regulate neurite outgrowth through binding to components of the extracellular matrix such as heparin and collagen I and IV. The splice isoforms that contain the BPTI domain possess protease inhibitor activity. Induces a AGER-dependent pathway that involves activation of p38 MAPK, resulting in internalization of amyloid-beta peptide and leading to mitochondrial dysfunction in cultured cortical neurons. Provides Cu(2+) ions for GPC1 which are required for release of nitric oxide (NO) and subsequent degradation of the heparan sulfate chains on GPC1.[32] [33] [34] [35] [36] Beta-amyloid peptides are lipophilic metal chelators with metal-reducing activity. Bind transient metals such as copper, zinc and iron. In vitro, can reduce Cu(2+) and Fe(3+) to Cu(+) and Fe(2+), respectively. Beta-amyloid 42 is a more effective reductant than beta-amyloid 40. Beta-amyloid peptides bind to lipoproteins and apolipoproteins E and J in the CSF and to HDL particles in plasma, inhibiting metal-catalyzed oxidation of lipoproteins. Beta-APP42 may activate mononuclear phagocytes in the brain and elicit inflammatory responses. Promotes both tau aggregation and TPK II-mediated phosphorylation. Interaction with Also bind GPC1 in lipid rafts.[37] [38] [39] [40] [41] Appicans elicit adhesion of neural cells to the extracellular matrix and may regulate neurite outgrowth in the brain (By similarity).[42] [43] [44] [45] [46] The gamma-CTF peptides as well as the caspase-cleaved peptides, including C31, are potent enhancers of neuronal apoptosis.[47] [48] [49] [50] [51] N-APP binds TNFRSF21 triggering caspase activation and degeneration of both neuronal cell bodies (via caspase-3) and axons (via caspase-6).[52] [53] [54] [55] [56] Evolutionary Conservation Check, as determined by ConSurfDB. You may read the explanation of the method and the full data available from ConSurf. Publication Abstract from PubMedThe major components of neuritic plaques found in Alzheimer disease (AD) are peptides known as amyloid beta-peptides (Abeta), which derive from the proteolitic cleavage of the amyloid precursor proteins. In vitro Abeta may undergo a conformational transition from a soluble form to aggregated, fibrillary beta-sheet structures, which seem to be neurotoxic. Alternatively, it has been suggested that an alpha-helical form can be involved in a process of membrane poration, which would then trigger cellular death. Conformational studies on these peptides in aqueous solution are complicated by their tendency to aggregate, and only recently NMR structures of Abeta-(1-40) and Abeta-(1-42) have been determined in aqueous trifluoroethanol or in SDS micelles. All these studies hint to the presence of two helical regions, connected through a flexible kink, but it proved difficult to determine the length and position of the helical stretches with accuracy and, most of all, to ascertain whether the kink region has a preferred conformation. In the search for a medium which could allow a more accurate structure determination, we performed an exhaustive solvent scan that showed a high propensity of Abeta-(1-42) to adopt helical conformations in aqueous solutions of fluorinated alcohols. The 3D NMR structure of Abeta-(1-42) shows two helical regions encompassing residues 8-25 and 28-38, connected by a regular type I beta-turn. The surprising similarity of this structure, as well as the sequence of the C-terminal moiety, with those of the fusion domain of influenza hemagglutinin suggests a direct mechanism of neurotoxicity. Solution structure of the Alzheimer amyloid beta-peptide (1-42) in an apolar microenvironment. Similarity with a virus fusion domain.,Crescenzi O, Tomaselli S, Guerrini R, Salvadori S, D'Ursi AM, Temussi PA, Picone D Eur J Biochem. 2002 Nov;269(22):5642-8. PMID:12423364[57] From MEDLINE®/PubMed®, a database of the U.S. National Library of Medicine. References
|
| ||||||||||||||||