3hf1: Difference between revisions
No edit summary |
No edit summary |
||
| (7 intermediate revisions by the same user not shown) | |||
| Line 1: | Line 1: | ||
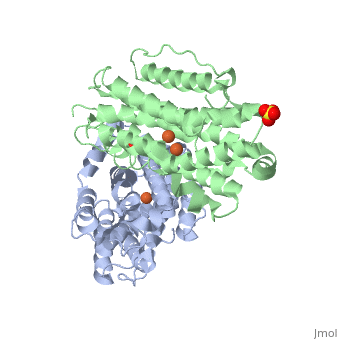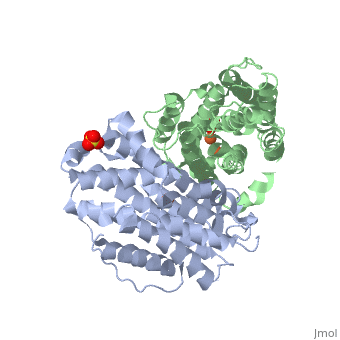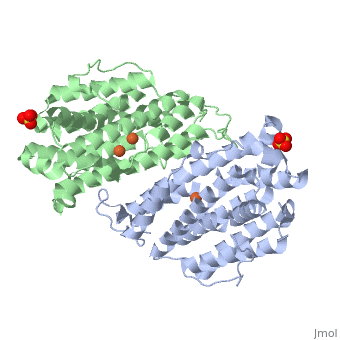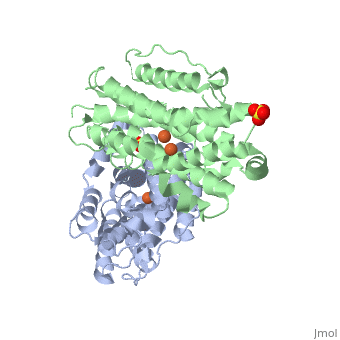
==Crystal structure of human p53R2== | |||
<StructureSection load='3hf1' size='340' side='right'caption='[[3hf1]], [[Resolution|resolution]] 2.60Å' scene=''> | |||
== Structural highlights == | |||
<table><tr><td colspan='2'>[[3hf1]] is a 2 chain structure with sequence from [https://en.wikipedia.org/wiki/Homo_sapiens Homo sapiens]. Full crystallographic information is available from [http://oca.weizmann.ac.il/oca-bin/ocashort?id=3HF1 OCA]. For a <b>guided tour on the structure components</b> use [https://proteopedia.org/fgij/fg.htm?mol=3HF1 FirstGlance]. <br> | |||
</td></tr><tr id='method'><td class="sblockLbl"><b>[[Empirical_models|Method:]]</b></td><td class="sblockDat" id="methodDat">X-ray diffraction, [[Resolution|Resolution]] 2.6Å</td></tr> | |||
<tr id='ligand'><td class="sblockLbl"><b>[[Ligand|Ligands:]]</b></td><td class="sblockDat" id="ligandDat"><scene name='pdbligand=FE:FE+(III)+ION'>FE</scene>, <scene name='pdbligand=SO4:SULFATE+ION'>SO4</scene></td></tr> | |||
<tr id='resources'><td class="sblockLbl"><b>Resources:</b></td><td class="sblockDat"><span class='plainlinks'>[https://proteopedia.org/fgij/fg.htm?mol=3hf1 FirstGlance], [http://oca.weizmann.ac.il/oca-bin/ocaids?id=3hf1 OCA], [https://pdbe.org/3hf1 PDBe], [https://www.rcsb.org/pdb/explore.do?structureId=3hf1 RCSB], [https://www.ebi.ac.uk/pdbsum/3hf1 PDBsum], [https://prosat.h-its.org/prosat/prosatexe?pdbcode=3hf1 ProSAT]</span></td></tr> | |||
</table> | |||
== Disease == | |||
[https://www.uniprot.org/uniprot/RIR2B_HUMAN RIR2B_HUMAN] Defects in RRM2B are the cause of mitochondrial DNA depletion syndrome type 8A (MTDPS8A) [MIM:[https://omim.org/entry/612075 612075]. A disorder due to mitochondrial dysfunction characterized by various combinations of neonatal hypotonia, neurological deterioration, respiratory distress, lactic acidosis, and renal tubulopathy.<ref>PMID:17486094</ref> <ref>PMID:18504129</ref> Defects in RRM2B are the cause of mitochondrial DNA depletion syndrome type 8B (MTDPS8B) [MIM:[https://omim.org/entry/612075 612075]. A disease due to mitochondrial dysfunction and characterized by ophthalmoplegia, ptosis, gastrointestinal dysmotility, cachexia, peripheral neuropathy. Defects in RRM2B are the cause of progressive external ophthalmoplegia with mitochondrial DNA deletions autosomal dominant type 5 (PEOA5) [MIM:[https://omim.org/entry/613077 613077]. A disorder characterized by progressive weakness of ocular muscles and levator muscle of the upper eyelid. In a minority of cases, it is associated with skeletal myopathy, which predominantly involves axial or proximal muscles and which causes abnormal fatigability and even permanent muscle weakness. Ragged-red fibers and atrophy are found on muscle biopsy. A large proportion of chronic ophthalmoplegias are associated with other symptoms, leading to a multisystemic pattern of this disease. Additional symptoms are variable, and may include cataracts, hearing loss, sensory axonal neuropathy, ataxia, depression, hypogonadism, and parkinsonism.<ref>PMID:19664747</ref> | |||
== Function == | |||
[https://www.uniprot.org/uniprot/RIR2B_HUMAN RIR2B_HUMAN] Plays a pivotal role in cell survival by repairing damaged DNA in a p53/TP53-dependent manner. Supplies deoxyribonucleotides for DNA repair in cells arrested at G1 or G2. Contains an iron-tyrosyl free radical center required for catalysis. Forms an active ribonucleotide reductase (RNR) complex with RRM1 which is expressed both in resting and proliferating cells in response to DNA damage.<ref>PMID:10716435</ref> <ref>PMID:11517226</ref> <ref>PMID:11719458</ref> | |||
== Evolutionary Conservation == | |||
[[Image:Consurf_key_small.gif|200px|right]] | |||
Check<jmol> | |||
<jmolCheckbox> | |||
<scriptWhenChecked>; select protein; define ~consurf_to_do selected; consurf_initial_scene = true; script "/wiki/ConSurf/hf/3hf1_consurf.spt"</scriptWhenChecked> | |||
<scriptWhenUnchecked>script /wiki/extensions/Proteopedia/spt/initialview01.spt</scriptWhenUnchecked> | |||
<text>to colour the structure by Evolutionary Conservation</text> | |||
</jmolCheckbox> | |||
</jmol>, as determined by [http://consurfdb.tau.ac.il/ ConSurfDB]. You may read the [[Conservation%2C_Evolutionary|explanation]] of the method and the full data available from [http://bental.tau.ac.il/new_ConSurfDB/main_output.php?pdb_ID=3hf1 ConSurf]. | |||
<div style="clear:both"></div> | |||
<div style="background-color:#fffaf0;"> | |||
== Publication Abstract from PubMed == | |||
Human p53R2 (hp53R2) is a 351 residue p53-inducible ribonucleotide reductase (RNR) small subunit. It shares >80% sequence identity with hRRM2, the small RNR subunit responsible for normal maintenance of the deoxyribonucleotide (dNTP) pool used for DNA replication, which is active during the S-phase in a cell-cycle dependent fashion. But rather than cyclic dNTP synthesis, hp53R2 has been shown to supply dNTPs for DNA repair to cells in G0-G1 in a p53-dependent fashion. The first x-ray crystal structure of hp53R2 is solved to 2.6 A, in which monomers A and B exhibit mono- and bi-nuclear iron occupancy, respectively. The pronounced structural differences at three regions between hp53R2 and hRRM2 highlight the possible regulatory role in iron assimilation, and help to explain previously observed physical and biochemical differences in the mobility and accessibility of the radical-iron center, as well as radical transfer pathways between the two enzymes. The sequence-structure-function correlations that differentiate hp53R2 and hRRM2 are revealed for the first time. Insight gained from this structural work will be used toward the identification of biological function, regulation mechanism and inhibitors selection in RNR small subunits. | |||
2.6 X-ray Crystal Structure of Human p53R2.,Smith P, Zhou B, Ho N, Yuan YC, Su L, Tsai SC, Yen Y Biochemistry. 2009 Sep 3. PMID:19728742<ref>PMID:19728742</ref> | |||
From MEDLINE®/PubMed®, a database of the U.S. National Library of Medicine.<br> | |||
</div> | |||
<div class="pdbe-citations 3hf1" style="background-color:#fffaf0;"></div> | |||
==See Also== | ==See Also== | ||
*[[Ribonucleotide reductase 3D structures|Ribonucleotide reductase 3D structures]] | |||
*[[Ribonucleotide reductase|Ribonucleotide reductase]] | == References == | ||
<references/> | |||
== | __TOC__ | ||
< | </StructureSection> | ||
[[Category: Homo sapiens]] | [[Category: Homo sapiens]] | ||
[[Category: | [[Category: Large Structures]] | ||
[[Category: Smith | [[Category: Smith P]] | ||
[[Category: Su | [[Category: Su L]] | ||
[[Category: Tsai | [[Category: Tsai S-C]] | ||
[[Category: Yen | [[Category: Yen Y]] | ||
[[Category: Yuan | [[Category: Yuan Y-C]] | ||
[[Category: Zhou | [[Category: Zhou B]] | ||
Latest revision as of 10:21, 6 September 2023
Crystal structure of human p53R2Crystal structure of human p53R2
Structural highlights
DiseaseRIR2B_HUMAN Defects in RRM2B are the cause of mitochondrial DNA depletion syndrome type 8A (MTDPS8A) [MIM:612075. A disorder due to mitochondrial dysfunction characterized by various combinations of neonatal hypotonia, neurological deterioration, respiratory distress, lactic acidosis, and renal tubulopathy.[1] [2] Defects in RRM2B are the cause of mitochondrial DNA depletion syndrome type 8B (MTDPS8B) [MIM:612075. A disease due to mitochondrial dysfunction and characterized by ophthalmoplegia, ptosis, gastrointestinal dysmotility, cachexia, peripheral neuropathy. Defects in RRM2B are the cause of progressive external ophthalmoplegia with mitochondrial DNA deletions autosomal dominant type 5 (PEOA5) [MIM:613077. A disorder characterized by progressive weakness of ocular muscles and levator muscle of the upper eyelid. In a minority of cases, it is associated with skeletal myopathy, which predominantly involves axial or proximal muscles and which causes abnormal fatigability and even permanent muscle weakness. Ragged-red fibers and atrophy are found on muscle biopsy. A large proportion of chronic ophthalmoplegias are associated with other symptoms, leading to a multisystemic pattern of this disease. Additional symptoms are variable, and may include cataracts, hearing loss, sensory axonal neuropathy, ataxia, depression, hypogonadism, and parkinsonism.[3] FunctionRIR2B_HUMAN Plays a pivotal role in cell survival by repairing damaged DNA in a p53/TP53-dependent manner. Supplies deoxyribonucleotides for DNA repair in cells arrested at G1 or G2. Contains an iron-tyrosyl free radical center required for catalysis. Forms an active ribonucleotide reductase (RNR) complex with RRM1 which is expressed both in resting and proliferating cells in response to DNA damage.[4] [5] [6] Evolutionary Conservation Check, as determined by ConSurfDB. You may read the explanation of the method and the full data available from ConSurf. Publication Abstract from PubMedHuman p53R2 (hp53R2) is a 351 residue p53-inducible ribonucleotide reductase (RNR) small subunit. It shares >80% sequence identity with hRRM2, the small RNR subunit responsible for normal maintenance of the deoxyribonucleotide (dNTP) pool used for DNA replication, which is active during the S-phase in a cell-cycle dependent fashion. But rather than cyclic dNTP synthesis, hp53R2 has been shown to supply dNTPs for DNA repair to cells in G0-G1 in a p53-dependent fashion. The first x-ray crystal structure of hp53R2 is solved to 2.6 A, in which monomers A and B exhibit mono- and bi-nuclear iron occupancy, respectively. The pronounced structural differences at three regions between hp53R2 and hRRM2 highlight the possible regulatory role in iron assimilation, and help to explain previously observed physical and biochemical differences in the mobility and accessibility of the radical-iron center, as well as radical transfer pathways between the two enzymes. The sequence-structure-function correlations that differentiate hp53R2 and hRRM2 are revealed for the first time. Insight gained from this structural work will be used toward the identification of biological function, regulation mechanism and inhibitors selection in RNR small subunits. 2.6 X-ray Crystal Structure of Human p53R2.,Smith P, Zhou B, Ho N, Yuan YC, Su L, Tsai SC, Yen Y Biochemistry. 2009 Sep 3. PMID:19728742[7] From MEDLINE®/PubMed®, a database of the U.S. National Library of Medicine. See AlsoReferences
|
| ||||||||||||||||||